
Abstract
Graphene oxide–polyaniline composites were synthesized by an interfacial method using two green solvents, water and an ionic liquid (1-butyl-3-methylimidazolium tetrafluoroborate), as the two phases. The interfacial polymerization of aniline was carried out at room temperature in the presence of graphene oxide dispersed in the aqueous phase. The analysis revealed the surface of the graphene sheets to be coated with a smooth thin layer of polyaniline. The thermal stability of the composites was much better than that of bare graphene oxide. The composites were used to modify the glass carbon electrodes for the chemical detection of hydrogen peroxide in aqueous media. This method is a facile, efficient, and green route for the development of doped polyaniline materials suitable for chemical sensors.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The conductivity of monolayer graphene changes as a function of the extent of surface adsorption, specific surface area, and Johnson noise [1, 2]. Many studies reported graphene to be a promising candidate for the detection of molecules ranging from gases to biomolecules [3–10]. Charge transfer between the adsorbed molecules and graphene is believed to be responsible for the chemical response. As molecules adsorb to the graphene surface, the location of adsorption experiences charge transfer with graphene as the donor or acceptor, which changes the Fermi level, carrier density, and electrical resistance of graphene.
Polyaniline (PANI) has attracted considerable attention for its unique and controllable chemical and electrical properties; environmental, thermal, and electrochemical stability; and interesting electronic, optical, and electro-optical properties [11–14]. PANI has a wide range of tunable properties emanating from its structural flexibility leading to potential applications in many fields, such as battery electrodes, anti-corrosive coatings, energy storage systems, gas sensors, and electrocatalytic devices [13, 14]. Moreover, PANI has the highest environmental stability and is recognized as the only conducting polymer stable in air [15]. PANI has been used as a sensor material because it is sensitive at room temperature and is expanding its application by combining with other nanomaterials. PANI has been used as a biosensor interface because it acts as an effective mediator for electron transfer in redox or enzymatic reactions and can also be used as a suitable matrix for the immobilization of biomolecules [16].
Composites consisting of graphene and PANI have been developed for different applications in lithium ion batteries, supercapacitors, catalysts, solar cells, nanodevices, chemical sensors, and biosensors [17–19]. Improved stability of the composites has been achieved due to the synergetic combination of the excellent conducting and mechanical properties of graphene sheets and the high pseudo-capacitance of PANI [20]. Moreover, the doping of graphene with polyaniline enhances the conductivity and mechanical strength remarkably, resulting in excellent performance as an electrode material for supercapacitors. In the sensor area, only a few studies on the use of graphene oxide (GO) and PANI composites as electrocatalysts have been reported. Fan et al. [21] fabricated an electrochemical sensor based on a graphene–polyaniline nanocomposite for the voltammetric determination of 4-aminophenol. This sensor showed favorable analytical performance for 4-AP detection with a low detection limit and high sensitivity. The GO/PANI composite was reported to show preferable electroactivity, even in neutral media, and exhibit excellent electrocatalytic activity in the simultaneous determination of ascorbic acid, dopamine, and uric acid [22]. On the other hand, preparing composite nanosheets with a homogeneous dispersion of individual graphene sheets within the smooth thin polymer layer is quite a challenge. This might reduce the electrocatalytic activity of the composites, limiting its further applications to electrochemical sensors.
This paper reports a simple and efficient method to synthesize GO/PANI composites by in situ interfacial polymerization using two green solvents, water and 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]). This method minimizes the use of organic solvents. The products obtained are characterized using the following techniques: X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), and ultraviolet–visible spectroscopy (UV–vis). The composites are also used to modify glass carbon electrodes for the chemical detection of hydrogen peroxide in aqueous media.
Experimental
Materials
Graphite powder (99.995 %, Alfa Aesar) was used as received. Aniline (99 %, Aldrich) was distilled under reduced pressure prior to use. [Bmim][BF4] (>98 %, Ionic Liquids Tech., Germany) was kept in a vacuum oven at 100 °C for 24 h to remove the volatile impurities prior to use. Other reagents were of analytical grade and used as received. Water was deionized in the laboratory.
Synthesis of GO
GO was synthesized from graphite powder using a modified Hummers method [23]. Typically, 2 g graphite powder was added to a 50-mL concentrated H2SO4 along with 2 g NaNO3 in a flask at 0 °C in an ice bath. Subsequently, 6 g KMnO4 was added slowly to the solution with vigorous stirring below 20 °C. The ice bath was removed and the flask was placed into another water bath at 35 °C, while keeping the mixture stirring for 3 h. The color of the resulting paste became brownish gray. The paste was diluted with 100 mL deionized water and stirred for 2 h. A 10-mL H2O2 (30 wt%) solution was added slowly to the suspension along with 100 mL of a HCl (10 % v/v) solution. The mixture was centrifuged and washed with deionized water until the decanted solution was neutral. The product was dried at room temperature under vacuum for 24 h.
Synthesis of GO/PANI composite
In a typical experiment, 0.5 g aniline was dissolved in 10 mL ionic liquid (IL) and 50 mg GO was dispersed in10 mL DI water, which was followed by the addition of 0.5 mL hydrochloric acid (1 M HCl) and 2.9 g potassium persulfate (KPS). The molar ratio of KPS to aniline was 2:1. The two solutions were transferred carefully to a vial and an interface was formed between the aqueous phase and IL phase. Green PANI formed at the interface was then diffused gradually into the aqueous phase. After 2 h, the entire water phase was filled homogeneously with a dark green GO/PANI nanocomposite. The aqueous phase was then collected and washed with ethanol and water to remove the unreacted chemicals and aniline oligomers. The GO/PANI composite obtained was dried in a vacuum oven at 40 °C for 24 h. As a comparison, pure PANI was prepared under the same conditions without adding GO in the aqueous phase.
Characterization
The samples were characterized by SEM (Hitachi, S-4200), TEM (Philips, CM200) at an acceleration voltage of 200 kV, XRD (PANalytical, X'Pert-PRO MPD) using Cu Kα radiation, and XPS (ULVAC-PHI, Quantera SXM) using an Al X-ray source. The FTIR (Excalibur Series FTS 3000, BioRad) spectra were recorded over 400–4,000 cm−1 at a resolution of 16 cm−1 within 32 scans using KBr pellets. UV–visible spectra were recorded over 200–800 nm using a UV–vis–NIR spectrophotometer (Cary 5000, Varian). Thermogravimetric analysis (TGA) was performed on a simultaneous TGA/DSC analyzer (SDT Q600, TA Instrument) from 30 to 600 °C at a heating rate of 10 °C/min under a nitrogen atmosphere.
All electrochemical experiments were performed on a VersaSTAT 3 AMETEK Model (Princeton Applied Research, USA) in a three-electrode configuration. Platinum foils and a saturated calomel electrode were used as the counter and reference electrodes, respectively. The working electrode was made using the same quantity of samples (GO, PANI, and GO–PANI) being coated onto carbon paper (2 × 2 cm in diameter) in all experiments. Typically, 50 mg sample was ultrasonicated in 5 ml N,N-dimethylformamide to form a homogenous dispersion. Subsequently, approximately 20 μl of the above solution was drop-cast onto the surface of a bare glassy carbon electrode and dried in an ambient atmosphere for 12 h.
Results and discussion
Figure 1 shows the preparation process of GO/PANI composites. The aniline monomer was dissolved in [bmim][BF4], and GO was dispersed in an aqueous acid solution with the oxidant, potassium persulfate. The water layer was spread carefully onto an equal volume of IL, forming a water/IL interface. After approximately 30 s, green polyaniline formed at the interface and gradually migrated to the aqueous phase. Finally, the entire water phase was filled homogeneously with a dark green GO/PANI composite, whereas the IL phase showed a red-orange color possibly due to the formation of aniline oligomers. The composite was collected and purified. Figure 1 also shows typical snapshots of the synthetic process in a water/IL system.

Schematic diagram of the synthesis of the GO/PANI composite and the progress of the interfacial polymerization of aniline in a water/IL system in the presence of GO
The surface morphology of the samples was examined by SEM and TEM. Figure 2 presents SEM images of raw graphite, bare GO, pure PANI, and GO/PANI. Graphite had a layered structure of stacked graphene sheets. GO showed thin sheets with distinct edges, wrinkled surfaces, and folding. A comparison with the composite morphology revealed a remarkable change in the physical nature of pure PANI after composite formation. The surfaces of the GO sheets were covered with PANI layers. To further characterize the composite morphology more clearly, bare GO, pure PANI, and GO/PANI composites with different resolutions were examined by TEM (Fig. 3). Sheets with wrinkled surfaces and folded edges were clearly visible in the image. The GO/PANI composites showed a different morphology compared to pure PANI due to the thin polymer layers covering the graphene sheets.

SEM images of graphite, bare GO, pure PANI, and GO/PANI composite

TEM images of the bare GO, pure PANI, and GO/PANI composite with different magnifications
The crystal structure of conducting PANI has a considerable effect on its electrical conductivity. A previous study reported that the highly ordered chain structure of conducting polymers could endow strong electrical conductivity [24]. Figure 4 shows the XRD pattern of graphite, GO, PANI, and GO/PANI composite. The graphite spectrum has a very strong peak at 26.5°; GO has a sharp peak at 11.85°. Pure PANI revealed a typical crystalline pattern, which was similar to that of the GO/PANI composite. The diffraction peaks at 21.17°, 23.70°, 25.43°, and 29.71° (= 2θ) with hkl values of (100), (110), (111), and (020), respectively, represent the emeraldine PANI [24, 25]. The peaks centered at 21.17° and 23.70° were attributed to the periodicity parallel and perpendicular, respectively, to the PANI chains. XRD results showed that PANI has a highly ordered crystal structure, which improves the crystallinity of the composite considerably, and is expected to exhibit high electrical conductivity. The decrease in peak height of the GO planes in the composite might be caused by the PAN polymer coating of the GO surface in the grafting process.

XRD patterns of graphite, bare GO, pure PANI, and GO/PANI composite
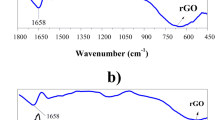
FTIR spectroscopy provided chemical structural information of GO, PANI, and GO/PANI composite (Fig. 5a). The spectrum of GO showed the representative peaks at 3,445 cm−1 (O–H), 1,721 cm−1 (C=O), and 1,042 cm−1 (C–O) [23]. The spectrum of PANI showed the usual characteristic stretching vibration bands at 1,561 cm−1 (C=C, quinoid rings), 1,492 cm−1 (C=C, benzenoid rings), 1,287 cm−1 (C–N), and 1,083 cm−1 (C–H). In the case of the GO–PAN composite, the FTIR spectrum was similar to that of PAN. There was no peak at 1,721 cm−1 because of C=O vibrations. These results confirmed that the surface of the GO was wrapped completely with PAN. The IR bands were in good agreement with previously reported data [24]. Figure 5b shows the TGA results of bare GO, pure PANI, and GO/PANI under a nitrogen atmosphere. All the samples showed similar decomposition curves. On the other hand, GO had much lower thermal stability than the pure PANI and GO/PANI composite, suggesting the pyrolysis of thermally oxygen-containing functional groups. The weight reduction of GO observed at temperatures less than 100°C and between 150 and 300 °C was consistent with the degradation of absorbed water and oxygen functional groups, respectively. These groups, such as −COOH and −OH, on the GO surface not only enhance the dispersibility, but can also anchor PANI to the surface [26]. The gradual weight loss of PANI between 100 and 280 °C was attributed to the deprotonation of PANI through the loss of dopant HCl. The major weight loss (∼30 %) before 600 °C was related to the degradation and decomposition of PANI with different degrees of polymerization. For the composite, the gradual degradation at approximately 280 °C was attributed to the evaporation of the absorbed solvent and the decomposition of oxygen groups on the GO surface. TGA suggested that the GO/PANI composite has higher and slightly lower thermal stability than GO and pure PANI, respectively. This might be due to the strong interactions between GO and PANI, which impose a restriction on the decomposition of the oxygen groups remaining on the GO surface. These interactions can result in homogeneous heating and the avoidance of heat concentrations [27].

a FTIR spectra of bare GO, pure PANI, and GO/PANI composite. b TGA thermograms of bare GO, pure PANI, and GO/PANI composite obtained with a ramping rate of 10 °C/min under a N2 atmosphere
Figure 6 showed the XPS survey spectra of the GO and GO/PANI composite as well as the core-level C 1s, N 1s, and O 1s peaks in the composite spectrum. The C 1s peak of GO could be deconvoluted to four peaks at binding energies (BEs) of 284.5 (C–C), 285.4 (C–OH), 286.8 (C–O–C), and 288.5 eV (C=O), respectively [28]. The spectra showed that the original GO sheets were partially oxidized and contained hydroxyl, carboxyl, and carbonyl groups. The core-level O 1s at a BE of 532.5 eV corresponded to the hydroxyl groups and surface adsorbed water. In the XPS spectrum of the GO/PANI composite, the peak representing nitrogen atoms was observed, indicating the presence of PANI. In the case of the composite, the intensity of both C 1s and O 1s peaks decreased after combination with PANI. The peaks corresponding to C–O–C (286.8 eV) and C–OH (285.4 eV) of graphene oxide disappeared. This was probably due to the increased conjugation and hydrogen bonding between the PANI backbone and graphene oxide sheets [26]. Remarkably, the new π→π* peak at approximately 293.3 eV appeared after polymerization, which was a characteristic of aromatic or conjugated systems, and it indicated an increased conjugation in the composites [28].

XPS survey spectra and core levels of C 1s, O 1s, and N 1s
The UV–vis absorption spectra of pure PANI, bare GO, and GO/PANI dispersed in ethanol were recorded (Fig. 7). The spectrum of GO revealed two absorption bands at 235 and 304 nm due to excitation of the π–π* transition of aromatic C–C and the n–π transition of C=O [29], respectively. The PANI spectrum revealed a sharp peak at 207 nm, which was assigned to the π–π* transition in the molecule conjugation. A weak peak at 265 nm and a broad peak at approximately 415 nm originate from the charged cationic species, which are known as polarons [30]. For the GO/PANI composite, two peaks were observed at 206 and 268 nm due to a π–π interaction between PANI chains and GO sheets in the composite [26]. Compared to PANI, the peak shift from 415 to 384 nm indicates that PANI is protonated in the synthesized composite. The UV–vis absorption results confirmed the strong interaction between the PANI polymer and GO sheets, which are in agreement with a previous study [26].

UV–visible spectra of bare GO, pure PANI, and GO/PANI composite in ethanol
Figure 8a shows the cyclic voltammograms of the bare glassy carbon electrode (GCE) and the electrodes modified with pure PANI (PANI/GCE) and GO–PANI composite (GO–PANI/GCE) in the presence and absence of hydrogen peroxide (H2O2) in a 0.1-M phosphate buffer solution (PBS) with pH 7.0. In the case of GCE, no anodic or cathodic responses were observed in the presence and absence of H2O2. This suggests that GCE does not detect H2O2 at the potential measured. For the modified electrodes, both PANI/GCE and GO–PANI/GCE showed normal redox behavior with a pair of well-defined and quasi-reversible redox peaks in the absence of H2O2. After adding a small amount of H2O2, the redox peak currents and reduced oxidation potential increased significantly at two modified electrodes, which were attributed to their good electrocatalytic activity. On the other hand, in both cases, the intensity of the GO–PANI/GCE peaks was higher than that of the PANI-modified electrode. The net values of the oxidation peak current of H2O2 obtained at GO–PANI/GCE (82.26 μA) were approximately 5.7 and 1.2 times higher than that of bare GCE (14.53 μA) and PANI/GCE (65.92 μA), respectively. This shows that the presence of GO enhanced considerably the electrochemical activity of the PANI film and its detection of H2O2.

Cyclic voltammograms of bare glassy carbon electrode (GCE) and electrodes modified with the pure PANI (PANI/GCE) and GO–PANI composite (GO–PANI/GCE) in 0.1 M PBS at pH 7.0 a in the absence of H2O2 and b in the presence of 12 mM H2O2 at scan rate of 200 mV/s
The effect of the scan rate of the potential on the electrocatalytic ability of the modified composite electrode for the detection of H2O2 was also studied. Figure 9a shows cyclic voltammograms of GO–PANI/GCE in 0.1 M PBS at pH 7.0 in the presence of 12 mM H2O2 at various scan rates. The redox peak currents increased linearly with increasing scan rate from 25 to 500 mV/s. This suggests that the electrochemical reaction of H2O2 on the GO–PANI film is a surface-controlled process [30]. The sensitivity of the electrode-modified composite was examined by varying the H2O2 concentration. Figure 9b presents the cyclic voltammograms of GO–PANI/GCE in 0.1 M PBS pH 7.0. The inset in Fig. 9a revealed a linear increase in peak current with increasing H2O2 concentration within the range of 2–24 mM. The major cathodic response was extremely sensitive to the H2O2 concentration. Therefore, the GO/PANI-modified electrode provided high sensitivity, highlighting the potential applications for fabricating H2O2 involved sensors.

a Cyclic voltammograms of the electrode-modified GO–PANI composite with different scan rates in the presence of 12 mM H2O2 and b with different concentrations of H2O2: a’ 2 mM, b’ 6 mM, c’ 12 mM, d’ 16 mM, e’ 18 mM, and f’ 24 mM at scan rate of 100 mV/s in 0.1 M PBS at pH 7.0
Conclusions
A GO/PANI composite was prepared efficiently in an IL–water system via the typical interfacial polymerization of an aniline monomer. XRD confirmed the highly ordered chain structure of pure PANI and PANI in the composite. SEM and TEM indicated that the polymer layer almost completely covered the GO sheets. The resulting GO/PAN composite showed preferable electroactivity, even in neutral media, and displayed excellent electrocatalytic activity towards the oxidation of H2O2. Overall, this material is suitable for determining the H2O2 concentration. This experimental method is a facile, efficient, and green route for developing GO/PANI-based materials for chemical sensors.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Katsnelson MI, Grigorieva IV et al (2005) Two-dimensional gas of massless Dirac fermions in graphene. Nature 438:197–200
Zhang YB, Tan YW, Stormer HL, Kim P (2005) Experimental observation of the quantum Hall effect and Berry's phase in graphene. Nature 438:201–204
Dan YP, Lu Y, Kybert NJ, Luo JT, Johnson ATC (2009) Intrinsic response of graphene vapor sensors. Nano Lett 9:1472–1475
Robinson JT, Perkins FK, Snow ES, Wei ZQ, Sheehan PE (2008) Reduced graphene oxide molecular sensors. Nano Lett 8:3137–3140
Ohno Y, Maehashi K, Yamashiro Y, Matsumoto K (2009) Electrolyte-gated graphene field-effect transistors for detecting pH and protein adsorption. Nano Lett 9:3318–3322
Zhang K, Zhang WL, Choi HJ (2012) Facile fabrication of self-assembled PMMA/graphene oxide composite particles and their electroresponsive properties. Colloid Polym Sci. doi:10.1007/s00396-012-2814-8
Huang B, Li ZY, Liu ZR, Zhou G, Hao SG, Wu J, Gu BL, Duan WH (2008) Adsorption of gas molecules on graphene nanoribbons and its implication for nanoscale molecule sensor. J Phys Chem C 112:13442–13446
Zhang WL, Choi HJ (2012) Fabrication and electrorheology of graphene oxide/ionic N-substituted copolyaniline composite. Colloid Polym Sci. doi:10.1007/s00396-012-2872-y
Leenaerts O, Partoens B, Peeters FM (2008) Adsorption of H2O, NH3, CO, NO2, and NO on graphene: a first-principles study. Phys Rev B 77:125416–125422
Yang J, Burkinshaw SM, Zhou J, Monkman AP, Brown PJ (2003) Fabrication and characteristics of 2-acrylamino-2-methyl-1-propanesulfonic acid-doped polyaniline hollow fibers. Adv Mater 15:1081–1084
Alivisatos AP (1996) Semiconductor clusters, nanocrystals, and quantum dots. Science 271:933–937
Dhand C, Das M, Datta M, Malhotra BD (2011) Recent advances in polyaniline based biosensors. Biosens Bioelectron 26:2811–2821
Bhadra S, Khastgir D, Singha NK, Lee JH (2009) Progress in preparation, processing and applications of polyaniline. Prog Polym Sci 34:783–810
Sergeyeva TA, Lavrik NV, Piletsky SA, Rachkov AE, Elskaya AV (1996) Polyaniline label-based conductometric sensor for IgG detection. Sens Actuat B-Chem 34:283–288
Luo YC, Do JS (2004) Urea biosensor based on PANi(urease)-Nafion/Au composite electrode. Biosens Bioelectron 20:15–23
Wang Y, Li Z, Wang J, Li J, Lin Y (2011) Graphene and graphene oxide: biofunctionalization and applications in biotechnology. Trends Biotechnol 29:205–212
Huang X, Yin Z, Wu S, Qi X, He Q, Zhang Q et al (2011) Graphene-based materials: synthesis, characterization, properties, and applications. Small 7:1876–1902
Zhou X, Wu T, Hu B, Yang G, Han B (2010) Synthesis of graphene/polyaniline composite nanosheets mediated by polymerized ionic liquid. Chem Commun 46:3663–3665
Huang YF, Lin CW (2012) Polyaniline-intercalated graphene oxide sheet and its transition to a nanotube through a self-curling process. Polymer 53:1079–1085
Fan Y, Liu JH, Yang CP, Yu M, Liu P (2011) Graphene–polyaniline composite film modified electrode for voltammetric determination of 4-aminophenol. Sens Actuat B-Chem 157:669–674
Bao Y, Song J, Mao Y, Han D, Yang F, Niu L et al (2011) Graphene oxide-templated polyaniline microsheets toward simultaneous electrochemical determination of AA/DA/UA. Electroanalysis 23:878–884
Nguyen VH, Kim BK, Jo YL, Shim JJ (2012) Preparation and antibacterial activity of silver nanoparticles-decorated graphene composites. J Supercrit Fluid 72:28–35
Tang Q, Wu J, Sun H, Lin J, Fan S, Hu D (2008) Polyaniline/polyacrylamide conducting composite hydrogel with a porous structure. Carbohyd Polym 74:215–219
Pouget JP, Jozefowica ME, Epstein AJ, Tang X, MacDiarmid AG (1991) X-ray structure of polyaniline. Macromolecules 24:779–789
Wang H, Hao Q, Yang X, Lu L, Wang X (2010) Effect of graphene oxide on the properties of its composite with polyaniline. ACS Appl Mater Inter 2:821–828
Hu H, Wang X, Wang J, Wan L, Liu F, Zheng H et al (2010) Preparation and properties of graphene nanosheets–polystyrene nanocomposites via in situ emulsion polymerization. Chem Phys Lett 484:247–253
Fan X, Peng W, Li Y, Li X, Wang S, Zhang G et al (2008) Deoxygenation of exfoliated graphite oxide under alkaline conditions: a green route to graphene preparation. Adv Mater 20:4490–4493
Chitara B, Krupanidhi SB, Rao CNR (2011) Solution processed reduced graphene oxide ultraviolet detector. Appl Phys Lett 99:113114–113116
Stejskal J, Kratochvil P, Radhakrishnan N (1993) Polyaniline dispersions 2. UV–Vis absorption spectra. Synth Met 61:225–231
Laviron E (1979) General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanal Chem Interfac 101:19–28
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2012009529).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nguyen, V.H., Tang, L. & Shim, JJ. Electrochemical property of graphene oxide/polyaniline composite prepared by in situ interfacial polymerization. Colloid Polym Sci 291, 2237–2243 (2013). https://doi.org/10.1007/s00396-013-2940-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-013-2940-y