
Abstract
The photochemical behavior of photosensitive materials obtained by spin-coating or network synthesis of a cellulosic polymer bearing photo-cross-linkable cinnamate groups was investigated. First, cinnamate groups were grafted on a cellulose acetate butyrate polymer, with different grafting densities. The photochemical properties of the polymers were studied in solution by UV–visible and 1H NMR spectroscopy. Then spin-coated films and networks were prepared and characterized as a function of the number of cinnamate groups per cellulosic unit. The water-wetting properties of both surfaces were studied by dynamic contact angle measurements, before and after photoirradiation, and subsequent heating. The surfaces obtained by the two methods have significantly different behaviors that can be assigned to the distinct photochemical pathways of the cinnamate groups upon irradiation depending on the sample preparation. Indeed, dimerization reaction is evidenced as the main process in the spin-coated films while the expected isomerization is predominant at the surface of the polymer networks.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Photosensitive polymers are extensively used in many practical applications such as photoresists, optical memory devices, or liquid crystal display panels [1].
The most studied photo-responsive polymer surfaces are those containing azobenzene groups [2–4]. As an example, the photochemical trans–cis isomerization that takes place under linearly polarized UV light has been extensively studied as an alternative method to the rubbing process for the photo-alignment of liquid crystals. In addition, the azobenzene trans–cis isomerizable polymer surfaces also attract considerable interest in the field of elaboration of wettability tunable surfaces [5, 6]. Indeed, the conformational transition induced by UV light is accompanied by a change in polarity leading to contact angle variations. Moreover, the cis–trans back isomerization can take place upon exposure to visible light, allowing reversible wetting modulation. It has also been shown that these wettability changes can be emphasized through surface texturation [7]. As a result, light appears as one of the promising driving forces to generate a gradient in surface energy allowing tuning surface wettability and thus manipulating liquid droplets on a surface. Indeed, the droplet motion can be guided spatially using controlled photoirradiation. Liquid motion on surfaces displaying a wettability gradient has also been investigated from a theoretical point of view [8].
Among photosensitive groups, photodimerizable chromophores such as cinnamate have also been investigated. Cinnamate groups can undergo both dimerization and trans–cis isomerization under UV light irradiation, depending on their local concentration. Their ability to cross-link is related to the carbon–carbon double bonds which can undergo a [2 + 2] cycloaddition reaction. Several applications take advantage of the irreversible dimerization reaction [9]. Indeed, polymers bearing cinnamate residues are among the most widely studied of photo-alignment materials for liquid crystal display [10–14]. The irreversible dimerization which takes place upon photoirradiation with linearly polarized UV light ensures homogeneous liquid crystal alignment. Cinnamate-modified polymer materials have also been used as optical wave-guide structures [15]. In this case, the photodimerization induces a decrease in the refractive index of the material, allowing photo-writing of an optical channel into the polymer film. Finally, to the best of our knowledge, the wettability changes induced by UV photoirradiation of cinnamate-modified polymer surfaces have not been investigated, unlike those of azobenzene isomerizable polymers.
In view of the design of photofunctional materials, the sample preparation can be a crucial factor in controlling the photochemical reaction processes which take place in the material. Indeed, morphological changes can be induced by the photochemical transformations, as a result of the geometrical changes of the photosensitive groups, and can affect the properties of the material [16, 17]. It has been shown that the azobenzene trans–cis isomerization implies a significant change in the section of the molecule. As a result, if the photosensitive groups are too close to each other, the conformation change can either be prevented or lead to an increase in surface roughness. In the case of photo-cross-linkable groups such as cinnamate, the dimerization needed for some applications can only take place if the distance between the reacting groups is appropriate. The flexibility of the cinnamate side chain may also play a role in the photochemical process [11]. These examples indicate that the elaboration method of photosensitive polymer surfaces can play a significant role on both the photochemical behavior of the material and thus its expected properties. Many strategies have been developed to design photosensitive polymer surfaces, including self-assembly [18], spin-coating [16], Langmuir–Blodgett technique [15, 19], and grafting of the polymer onto the surface [20]. Polymer network materials could be another interesting route to obtain photosensitive surfaces. Such materials can be obtained from the use of either a photo-cross-linkable polymer or a photosensitive polymer cross-linked by an exogenous agent. The photoreactivity of polymers bearing cinnamate groups can thus be exploited to obtain cross-linked materials, i.e., photosensitive polymer networks that show many advantages [21]. Indeed, cross-linking presents the advantage of ensuring the stability of both bulk and surface morphology, even upon heating. In addition, in view of contact angle measurements with different liquids, cross-linking restricts the solubility of the material in organic solvents [22].
In the present work, the photochemical behavior of cinnamate-modified polymer materials obtained from either spin-coating or polymer network synthesis was compared. The evidenced photochemical processes were then connected to the wetting behavior of water on the surfaces. First, cinnamate groups were grafted on a cellulose acetate butyrate (CAB) polymer with different substitution degrees. The photochemical properties of these modified polymers (CABg) were followed in solution by means of UV–visible and 1H NMR spectroscopies. Then, the wetting behavior of water on spin-coated films of CAB and CABg was investigated upon photoirradiation of the surfaces and correlated to UV absorption characterizations. Finally, polymer networks based on CAB or CABg were synthesized. The thermomechanical properties of the networks containing different proportions of cinnamate groups were analyzed by dynamic mechanical thermal analysis (DMTA). The wetting properties of water on the CAB or CABg networks synthesized with different substitution degrees were investigated and compared with those of the corresponding spin-coated films.
Experimental section
Materials
CAB (6 mol% acetate, 77 mol% butyrate, and 17 mol% hydroxyl; Mn = 20,000 g mol−1, Acros), cinnamoyl chloride (Acros), dibutyltin dilaurate (DBTDL, Aldrich), triethylamine (Aldrich), poly(ethylene glycol) dimethacrylate (Mn = 330 g mol−1, Aldrich), and Desmodur® N3300 (NCO content by weight of 21.8 ± 0.3% according to the supplier, Bayer) were used as received. 2,2-Azobisisobutyronitrile (Acros) was recrystallized in methanol before use. Chloroform and dichloromethane (puro) were purchased from Carlo Erba.
Synthesis of CABg
CAB was modified as follows: 1.0 g CAB and 0.2 mL triethylamine were dissolved in 10-mL anhydrous dichloromethane then 0.25 g of cinnamoyl chloride was added dropwise. The mixture was kept at reflux under stirring. After a given time depending on the expected substitution degree, the reaction mixture was washed with water to remove the triethylammonium chloride. Dichloromethane was then removed by rotary evaporation. The remaining white solid was purified by Soxhlet extraction for 7 days with hexane and then dried under vacuum. CABg with different number of cinnamate groups per cellulosic unit (ds*) up to 0.25 were prepared using different reaction times. CABg with a ds* of 0.39 was obtained by repeating the 96 h reaction once more [23]. ds* was determined by 1H NMR spectroscopy.
Spin-coating of CAB and CABg
A 2 wt.% chloroform solution of CAB or CABg was spin-coated onto glass substrates at 6,000 rpm for 60 s. Prior to use, the substrates were carefully cleaned in a beaker of ultra-pure water (18 MΩ cm Millipore) placed in an ultrasonic bath for at least 20 min at room temperature and were then dried with argon stream. Under these conditions, a 60-nm thick film was obtained, as measured with a DEKTAK 150 profilometer.
Synthesis of CAB and CABg networks
The cellulosic network was prepared by condensation of CAB or CABg (1 g) with Desmodur® N3300 (0.25 g), in 8 mL of dichloromethane, in the presence of DBTDL (3·10−8 mol–18 μL) as catalyst [24]. The mixture was poured in a Petri dish, and the solvent was allowed to evaporate at room temperature for 12 h during which the reaction proceeds slowly.
A particular attention was paid to the Petri dishes cleaning since the contact angle measurements were performed on the network surface in contact with the glass. Thus, Petri dishes were carefully cleaned with sulfochromic acid and then rinsed with ultra-pure water (18 MΩ cm, Millipore).
Analytical techniques
The polymer molecular weights were determined by steric exclusion chromatography (Waters) equipped with Stryragel HR columns, a model 486 absorbance detector and a model 410 differential refractometer. The retention times were recorded at a flow rate of 1.0 mL min−1 with THF as eluent. The molecular weights were determined by fitting of a standard polystyrene calibration curve.
1H NMR spectra were recorded on a Bruker Avance DPX 250 NMR spectrometer. The compounds were dissolved in deuterated chloroform (Aldrich).
The UV–visible spectra were recorded on a V570 spectrometer (JASCO®). The dilute (10−4 mol L−1) chloroform solutions were analyzed in a Suprasil® cell with a 1-cm path.
In order to determine the amount of unreacted starting materials in the final materials, networks were extracted for 48 h in a Soxhlet with dichloromethane. After extraction, the sample was dried under vacuum and then weighed. The soluble fraction is given as a weight percentage: \( \frac{{{W_0} - {W_E}}}{{{W_0}}} \cdot 100 \) where W 0 and W E are the weights of samples before and after extraction, respectively.
DMTA measurements were carried out on network samples with a Q800 apparatus (TA Instruments) operating in tension mode with a 10-nN pre-stress. Experiments were performed at a frequency of 1 Hz and a heating rate of 3 °C min−1 from −70 to 220 °C. Typical sample dimensions were 20 × 5 × 1 mm. The set-up provides the storage and loss moduli (E′ and E″) and the damping parameter or loss factor (tan δ).
The atomic force microscopy (AFM) experiments were performed with a Nanoscope IIIA Dimension 3100 microscope from Digital Instruments. Measurements were carried out in air at room temperature. The images were obtained with the tapping mode and a 256 × 256 dots resolution. For each sample, AFM images were recorded on at least four samples and in several places in order to check the reproducibility of the displayed images.
Photoirradiation was carried out using a high-pressure Hg lamp (100 W, Lot-Oriel) equipped with a glass filter (λ > 280 nm). For contact angle studies, the samples were illuminated 4 h at a distance of 50 cm with the lamp power set to 80 W.
All contact angle measurements were carried out under air at 20 °C. Advancing and receding contact angles of water were determined using the drop shape analysis profile device equipped with a tiltable plane (DSA-P; Kruss, Germany). A 40-μL ultra-pure water drop was first deposited on the substrate using a variable volume micropipette. The static corresponding contact angle was measured by means of a Young–Laplace drop profile fitting. The water surface tension was also deduced in order to check systematically that no water contamination by polymer residues occurs during polymer network exposure. In order to perform dynamic contact angle measurements, the solid surface sustaining the drop was then tilted at a constant speed (2°/s) and the images of the drop simultaneously recorded. The advancing and receding angles were measured respectively at the front edge and the rear edge of the drop, just before the triple line starts moving. The angles were obtained using the tangent of the drop profile at the triple line.
For each network or spin-coated film, contact angles were measured on about five samples resulting from different syntheses; six drops per sample were analyzed. The reported contact angle values correspond to the average of all measurements with an error bar corresponding to the standard deviation.
Results and discussion
Native cellulose is highly crystalline and thus totally insoluble in common solvents. However, acetate, butyrate, and acetate butyrate substituted celluloses are thermoplastic polymers which can be amorphous and soluble in common solvents according to the degree of hydroxyl modification [25]. Among cellulose derivatives, ester-modified celluloses such as CAB are very good film-forming materials with high scratch resistance. Commercial CAB is available with free OH content between 4 and 17 mol%. The choice of starting CAB material was crucial in this study. Indeed, a significant proportion of hydroxyl functions is necessary to allow the chromophore grafting in the one hand, and, the cross-linking reaction leading to the network formation, on the other hand. Moreover, the polymer must be sufficiently modified compared with native cellulose to be soluble in organic solvents. The substituted CAB used in this work contains 17 mol% free OH corresponding to an average of 0.5 OH group per cellulosic unit (Fig. 1).

1H NMR spectra recorded on CDCl3 solutions of CAB (bottom) and cinnamate-modified CABg (top) corresponding to a ds* of 0.25
First, the grafting of cinnamate groups on CAB is described and the CABg solutions is investigated. Second, spin-coated films of CAB and CABg are studied by means of water contact angle measurements before and after photoirradiation. The results are correlated to UV absorption characterizations. Finally, the wetting properties of these thin films are compared with those of polymer networks of CAB and CABg.
Synthesis and characterization of photosensitive CAB polymers
Cinnamate groups were grafted on a CAB by addition reaction between cinnamoyl chloride and CAB free hydroxyl functions. The reaction was carried out in refluxing anhydrous dichloromethane. Different reaction times were used in order to vary the density of cinnamate groups. Thus, the CABg obtained for different reaction times were characterized by steric exclusion chromatography (SEC) while the number of cinnamate groups per cellulosic unit (ds*) was determined by 1H NMR spectroscopy.
Figure 1 presents the 1H NMR spectra obtained for deuterated chloroform solutions of CAB and CABg corresponding to a ds* of 0.25, as a typical example. Three different regions can be distinguished on the CABg spectrum. The protons of the acetate and butyrate groups are detected between 0.5 and 2.5 ppm (C region). The protons of the cellulosic pattern are located between 2.7 and 5.3 ppm (B region) while the cinnamate group protons are detected between 6.0 and 8.0 ppm (in addition to the chloroform peak at 7.27 ppm—A region). The detection of the aromatic protons confirms the presence of cinnamate groups grafted on the CAB polymer. The number of cinnamate groups per cellulosic unit, denoted as ds*, is calculated as the ratio of the number of protons in the cinnamate group (7H by unit) and the number of protons in the cellulosic ring (7H by unit also). Various ds* were obtained depending on the reaction time (Table 1). Thus, ds* reaches its maximum value, 0.25 ± 0.01, for a 96-h reaction time. In order to obtain a higher grafting density, the 96-h reaction was carried out once more on the ds* = 0.25 modified sample, leading to a ds* of 0.39 ± 0.01.
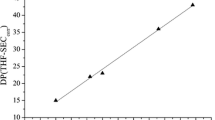
Chromatograms recorded on CAB and CABg solutions are reported in Fig. 2. The same retention time is observed, meaning that no significant depolymerization occurs during the grafting reaction. In order to check that cinnamate groups are well grafted on CAB, two different detectors were used: a differential refractometer which allows the CAB detection and an absorbance detector (λ = 270 nm) exclusively sensitive to cinnamate groups.

Chromatograms recorded with refractometer (circles, right scale) and UV (solid line, left scale) detectors on THF solutions of CAB (a) and cinnamate-grafted CAB corresponding to a ds* = 0.25 (b). For clarity, the different curves were shifted
On the one hand, no UV signal close to the solvent retention time is detected (Fig. 2b), ensuring that cinnamoyl chloride or cinnamic acid from the starting material has been removed efficiently by the sohxlet extraction during the purification process. On the other hand, the signal measured at 270 nm with the UV detector on the CABg solutions corresponds exactly to the retention time of the unmodified CAB measured with the differential refractometer. Cinnamate groups are thus well grafted on CAB.
These 1H NMR and SEC characterizations show that the proportion of grafted groups can be easily controlled by the reaction time (Table 1) and confirm that cinnamate groups are efficiently grafted on CAB polymer.
Then, the photochemical behavior of CAB modified with cinnamate groups (CABg) was followed by monitoring UV–visible spectra of dilute chloroform solutions. First, UV–visible spectra of chloroform solutions of cinnamoyl chloride and CABg were recorded. The CABg solution shows a maximum absorbance at 282 nm while the maximum absorbance is detected at 301 nm for the cinnamoyl chloride solution. The observed hypsochrome shift confirms that cinnamate groups are well grafted on the CAB polymer. Second, the molar extinction coefficient of the grafted cinnamate group can be determined from these UV spectra according to the Beer-Lambert law. The molar extinction coefficient of the trans isomer was thus estimated to about 14,000 L mol−1 cm−1 at 282 nm in chloroform, in good agreement with the value reported by Ichimura et al. [11].
CABg solutions were then submitted to different times of photoirradiation. Figure 3a shows the UV absorption spectra measured before and after photoirradiation while the maximum absorbance at 282 nm is plotted as a function of time on Fig. 3b. A gradual decrease of the absorbance is observed and a stationary state is reached after about 20 min. In addition, as shown in the inset of Fig. 3a, b, an isosbestic point is evidenced at 317 nm. The observation of an isosbestic point within the studied range of photoirradiation times indicates that the trans to cis isomerization is the predominant process [11]. Thus, assuming that the dimerization reaction is negligible, and that the molar extinction coefficient of the cis isomer is negligible with respect to the one of the trans isomer, as reported by Ruslim et al. [26], the proportion of trans and cis isomers can be estimated from the decrease in the absorption peak maximum as being 63% and 37%, respectively, after a 60-min photoirradiation of the solution.

a UV–visible spectra of a 2·10−5-mol L−1 CABg chloroform solution before photoirradiation and after different times of photoirradiation (t = 2, 4, 6, 10, 20, 30, and 50 min) and inset: zoom of the isosbectic point; b variations of the maximum absorbance at 282 nm (squares, left scale) and of the absorbance at 317 nm (circles, right scale) as a function of time
In order to check that no dimerization reaction occurs as expected in dilute solution [27], 1H NMR spectra were monitored before and after photoirradiation (Fig. 4a, b). It should be emphasized that, for a better detection, the 1H NMR solutions were more concentrated (10−1 mol L−1) than the solutions used for UV–visible experiments (10−4 mol L−1). Dimerization reaction was thus more likely to occur under the 1H NMR characterization conditions than during the previously reported photoirradiation. The chemical shifts of the α-vinylic H are assigned at 6.4 and 5.9 ppm for the trans and cis isomers, respectively [28]. The chemical shift associated to the β-vinyl H being located at 7.8 and 7.0 ppm for the trans and cis forms, respectively, i.e., in the region of the aromatic protons, the modification shift of the β-vinyl protons cannot be followed upon photoirradiation.

1H NMR spectra recorded on a CDCl3 CABg solution (ds* = 0.25), a before and b after 4 h photoirradiation, and c then 16 h heating at 55 °C
The fraction of trans (respectively cis) isomer was calculated from the ratio between the peak integral corresponding to the α-vinyl H of the trans (respectively cis) form at 6.4 ppm (respectively 5.9 ppm) and the sum of the peak integrals corresponding to the whole α-vinyl H at 6.4 and 5.9 ppm. Before irradiation, only protons characteristic of the trans isomer are detected at 6.4 ppm. After 4 h irradiation, protons characteristic of the trans (68%, 6.4 ppm) and cis (32%, 5.9 ppm) isomers are evidenced indicating that the CABg polymer undergoes a partial trans–cis isomerization upon irradiation. The fraction of cis isomer is in good agreement with the one previously estimated from the decrease in the UV absorption peak.
The chemical shift related to dimer formation would be observed at 3.8 and 4.4 ppm [29], i.e., in the same region as the cellulosic protons. Thus, it is not possible to detect dimers directly. However, the sum of the peak integrals corresponding to the trans and cis α-vinyl H, normalized to the peak integral corresponding to the cellulosic protons remains constant before and after photoirradiation. Therefore, no dimer formation occurs even though the solution concentration is higher under NMR conditions.
As the trans–cis isomerization is the predominant process, a cis to trans switching should be possible. Unlike azobenzene compounds, there is no suitable wavelength for irradiation at which cis isomer is preferentially excited in order to obtain the reverse cis–trans isomerization. As cinnamate derivatives are mainly investigated due to their potential applications in liquid crystals display technology, the cis to trans isomerization process is scarcely described in the literature. However, it can be thermally activated [30]. That is why the 0.5-h irradiated CABg solution was then heated at 55 °C, just below the chloroform boiling point. The UV absorption spectrum monitored after photoirradiation and 1 h heating shows that the absorbance increases slightly but does not recover its initial value (Fig. 5). Indeed, from the variation in maximum absorbance, only 14% of the cis isomers have recovered the trans form. Due to the sensitivity of the absorbance to changes in solution concentration, it was difficult to apply longer heating time. The cis to trans switching was then studied by 1H NMR with varying heating time. Even upon 16 h heating, only 76% of cinnamate groups retrieved their initial trans state (Fig. 4c). As no dimer formation occurs, this could be due to the heating conditions, either too low temperature or too short heating time. Indeed, Hocking reported a complete cis to trans isomerization upon heating at 143 °C for several days [28]. Higher temperatures are probably required to obtain a complete reversibility. These results show that, in the range of studied concentrations (<10−4 mol L−1), cinnamate groups grafted on a CAB polymer exclusively undergo a trans–cis isomerization. However, due to the low boiling point of chloroform, the cis to trans isomerization is only partial. Such limitation might be avoided in the case of the CABg spin-coated films or polymer networks, i.e., free-solvent systems, which can be heated up to higher temperatures.

UV–visible spectrum of a CABg solution a before photoirradiation, b after 0.5-h photoirradiation, and c and after 0.5-h photoirradiation followed by 1 h heating at 55 °C
Spin-coated CAB films
CAB and CABg polymers were spin-coated on glass substrates in order to study their wetting behavior upon photoirradiation. CABg bearing two different numbers of cinnamate groups per cellulosic unit, ds* = 0.16 and 0.39, were used.
AFM images obtained on CABg films (Electronic supplementary material (ESM)) show no effect of the irradiation on the surface roughness, which remains close to 1 nm.
The advancing and receding contact angles of water measured before (θ a, θ r) and after (θ *a , θ *r ) photoirradiation are presented in Table 2. The variations upon irradiation(θ a–θ *a ) and (θ r–θ *r ) are plotted in Fig. 6. Advancing contact angle values measured on the films before irradiation do not vary much with increasing ds*, in the range 0–0.39. The receding contact angle is a few degrees lower for the unmodified CAB samples than for the CABg ones. Even if the root mean square roughness measured on 10 × 10 μm as well as on 1 × 1 μm is close to 1 nm on the whole surfaces, some holes, 30 nm deep, were detected on the unmodified CAB, that could result in a higher contact angle hysteresis (Fig. S1 in the ESM). Upon photoirradiation, the advancing and receding contact angles do not vary for the CAB films, in agreement with the absence of cinnamate groups in the polymer. For the CABg films, the irradiation has no significant effect on the advancing contact angle while it leads to a few degrees increase of the receding contact angle, for the two probed ds*.

Variation, upon photoirradiation, of the advancing a and receding b water contact angles measured on CABg spin-coated films with different number of cinnamate groups per cellulosic unit (ds*): a θ a*–θ a; b θ r *–θ r. Images of droplets obtained on a CABg spin-coated film (ds* = 0.39) before (left) and after (right) photoirradiation illustrate the advancing contact angle measurement
Contact angle variations upon photoirradiation of photosensitive organic surfaces were mainly reported for azobenzene-modified polymer surfaces obtained by grafting, spin-coating or self-assembly. They all evidence a few degree decrease of either the static or the advancing water contact angle attributed to the change in dipole moment resulting from the trans to cis isomerization [16, 27, 31–33]. In some cases, due to an increase in surface roughness upon irradiation or to the film preparation on a rough substrate, an enhancement of contact angle variations is detected [7, 17, 34]. Fewer studies were devoted to the variation of the receding contact angle upon irradiation [3, 16, 30]. Both few degree increasing and decreasing values were measured after irradiation depending on the azobenzene-modified polymer and also on the probe liquid.
The isomerization thus manifests itself by the decrease in the dipole moment of the photosensitive group leading to a decrease in static or advancing contact angle. The absence of significant variation of θ a upon irradiation of the spin-coated films could be thus the result of photodimerization of the cinnamate groups.
In order to check this hypothesis, two experiments were carried out. On one hand, the different irradiated spin-coated films were dipped into chloroform. While the film of the non grafted polymer dissolves completely in chloroform, the CABg films are found to be insoluble. This means that an exogenous cross-linking reaction of the polymer chains occurred. In the absence of exogenous cross-linker, the present reaction should be the dimerization of the cinnamate groups. On the other hand, UV absorption spectra of the CABg spin-coated films were measured before and after different irradiation times and then after 4 h irradiation followed by 0.5 h heating at 130 °C (Fig. 7). The absence of reversibility in such heating conditions should be indicative of dimerization. Before photoirradiation, the maximum absorbance is measured at 282 nm, nearly the same value as the one obtained for chloroform CABg solutions. This means that the interactions between cinnamate groups in the condensed films do not induce any modification of the absorption spectrum. Upon photoirradiation from 1 to 4 h, a gradual decrease of the maximum absorbance is observed in the same way as it occurs in solution. However, the inset of Fig. 7 shows that this decrease is accompanied by a deviation from the isosbestic point evidenced in solution at 317 nm. This deviation is indicative of a dimerization process [11]. The 4 h irradiated film was then heated for 0.5 h at 130 °C. A subsequent decrease of the absorption peak is detected. In the case of a reversible cis to trans switching, an increase of the absorbance is expected, as observed in solution. The isomerization process is thus probably negligible. Such a decrease upon heating was previously reported by Sung et al. for spin-coated poly(vinyl cinnamate) and poly(3-ethylene-alter-1 vinyl cinnamate) films and was assigned to the occurrence of a thermal dimerization reaction between cinnamate groups [35]. In order to confirm that the dimerization reaction can be activated through heating in the same way as through photoirradiation, the UV absorption spectra of an as-prepared film were measured as a function of the heating time up to 1 h at 130 °C. The same decrease in the maximum absorbance as upon photoirradiation was detected. The dimerization reaction is thus favored by the increase in polymer chain mobility through heating. Finally, the disappearance of the isosbestic point upon photoirradiation associated to the irreversibility of the phenomenon indicates the formation of cinnamate dimers in the irradiated spin-coated CABg films.

UV–visible spectra of a CABg spin-coated film obtained for ds* = 0.39 before photoirradiation, after 1, 2, 3, and 4 h photoirradiation times, and after 4 h photoirradiation followed by 0.5 h heating at 130 °C (solid symbols). Inset: zoom showing the deviation from the isosbectic point
Therefore, both solubility experiments and UV absorption characterizations of the irradiated CABg spin-coated films confirm the dimerization of the cinnamate moieties as the measured process.
So the variation in advancing contact angles upon irradiation of the CABg spin-coated films could result from both photodimerization and isomerization of the cinnamate groups present at the surface. In addition, in case of dimerization, the process should not be reversible upon heating. The spin-coated films were thus heated 0.5 h at 130 °C after photoirradiation. AFM images obtained on CABg films (Fig. S1 in the ESM) show no effect of the heating process on the surface roughness. The measured advancing and receding contact angles (θ heata , θ heatr ) are reported in Table 2. It was checked that such irradiation followed by heating has no influence on the contact angle values for the unmodified CAB films. For the CABg films, one can notice that the receding contact angles do not retain their initial values. For the samples obtained with the CABg synthesized with the higher ds*, θ r keeps exactly the same value as after irradiation, meaning that the dimerization is predominant at the surface. For the films obtained with the CABg corresponding to ds* = 0.16, θ r slightly decreases toward its initial value, probably indicating both dimerization and isomerization occurrence at the surface.
In order to investigate whether dimensionally stable bulk materials could have an effect on the wetting behavior of water on CAB and CABg, a similar study on CAB and CABg polymer networks was carried out in which controlled polymer cross-linking densities were performed by an exogenous agent, i.e., a pluri-isocyanate compound.
CABg networks
First, the possible effect of photoirradiation and then heating on the wetting properties of water on CABg polymer networks synthesized with different densities of cinnamate groups (ds* = 0–0.39) was investigated.
As previously mentioned, cinnamate dimerization can occur in spin-coated films during photoirradiation. In order to check whether cinnamate dimerization occurs during the synthesis of CABg polymer networks, we tried to synthesize a CABg network exclusively from the cinnamate dimerization reaction, i.e., without any cross-linker agent. A dichloromethane CABg solution was thus poured in a Petri dish and 4 h photoirradiated until complete solvent evaporation. A soluble fraction of 100% was then measured by sohxlet extraction, meaning that no cross-linking at all occurred. This result shows that the dimerization can be considered as negligible under these synthesis conditions. The difference in photochemical behavior with respect to the spin-coated films in which dimerization occurs can be explained by the thickness of the materials, 60 nm for the films against 500 μm for the networks.
CABg networks were thus synthesized using an exogenous cross-linking agent. The synthesis was carried out by a condensation reaction between CABg free hydroxyl groups of the cellulose derivative and the isocyanate functions of a cross-linker (Desmodur® N3300). Since the number of free OH groups available for the cross-linking varies as function of ds*, it appeared crucial to characterize the role of the cinnamate grafting on the cross-linking quality and mechanical properties of the networks. Indeed, as previously mentioned, the CAB hydroxyl functions allow both the cinnamate groups grafting and the network cross-linking. Thus, as the surface properties of the networks could be influenced by their bulk characteristics, the CABg networks were first characterized by soxhlet extraction and DMTA. The bulk properties were then compared with those of the unmodified CAB networks.
The unreacted precursor content was determined by Soxhlet extraction with chloromethane and found to be low, close to 4%, for CAB and CABg networks obtained for different ds* ranging from 0.1 to 0.25. These whole CAB-based polymers can be thus considered as correctly cross-linked. In contrast, the networks synthesized with the more substituted CABg (ds* 0.39, i.e., 0.11 free OH by cellulosic unit), displaying a higher unreacted precursor content, close to 10%, were not considered for subsequent studies.
The storage and loss moduli of the different networks were then measured by means of DMTA. The plot of the loss tangent as a function of the temperature is shown in Fig. 8 for the non-modified CAB network and two CABg networks bearing 0.1 and 0.25 cinnamate groups per cellulosic unit, respectively. The storage modulus values are also reported.

Loss tangent (left scale) and storage modulus (right scale) of CAB network (squares) and CABg networks obtained for ds* = 0.1 (circles) and ds* = 0.25 (triangles)
The mechanical α-relaxation temperatures (T α) determined at the maximum tanδ value for the different networks are very close to each other. Indeed, the T α is obtained at 121 and 125 °C for the non-modified CAB and CABg networks, respectively. Nearly the same T α is thus measured for the CABg networks obtained for different ds* ranging from 0.1 to 0.25, meaning that the cross-linking density is comparable for all samples and that the grafted cinnamate units do not impair the CAB polymer chain cross-linking process. This is confirmed by dichloromethane swelling experiments leading to a maximum swelling ratio in between 145% and 155% for all samples. In addition, the storage modulus E′ at 20 °C provided by the DMTA set-up was shown to be close to 2,000 MPa, independently of ds* in the range 0–0.25.
In order to check the possible tuning of surface wettability by photoirradiation of the cinnamate groups, measurements of advancing and receding water contact angles were performed on CAB and CABg networks before and after irradiation (Table 3). Before this, the surface topography of the networks was imaged by means of AFM (Fig. S2 in the ESM) which shows no effect of the irradiation on the surface roughness, close to 1 nm.
First, before irradiation, the advancing contact angle values obtained on the networks synthesized with varying number of cinnamate groups show that the grafting only induces a slight increase in the hydrophobicity of the surfaces. Compared with the values measured on the spin-coated films, θ a is in the same range, even slightly lower. In contrast, θ r is about 20° lower for the networks, meaning that the contact angle hysteresis is strongly higher. The hysteresis can result from both surface roughness and chemical heterogeneities. The root mean square roughness measured on 10 × 10 μm as well as on 1 × 1 μm AFM images is of the same order on the two types of surfaces (Figs. S1 and S2 in the ESM). The difference in contact angle hysteresis could be due to a higher surface roughness of the networks at a smaller length scale than the probed one or to chemical heterogeneities arising from Desmodur® N3300. Indeed, in the networks, the CAB or CABg polymer is associated to a cross-linker which is absent in the spin-coated films.
The variations in advancing contact angle (θ a*–θ a) and receding contact angle (θ r*–θ r) upon irradiation are plotted in Fig. 9a, b, respectively. After irradiation, the contact angles measured on unmodified CAB networks keep the same values (81.7°), as expected in the absence of photosensitive groups. For CABg networks, the advancing contact angle decreases after irradiation, the variation being all the more important since ds* is high. In contrast, the receding contact angle increases after irradiation, in a more pronounced way with higher ds*.

Variation, upon photoirradiation, of the advancing (a) and receding (b) water contact angles measured on CABg networks with the number of cinnamate groups per cellulosic unit (ds*): a θ a* –θ a; b θ r *–θ r. Images of droplets obtained on a CABg network (ds* = 0.25) before (left) and after (right) photoirradiation illustrate the advancing contact angle measurement
The polymer networks have thus a quite different wetting behavior upon photoirradiation than the spin-coated films. Indeed, a decrease of the advancing contact angle is evidenced for the CABg networks whereas no variation was detected for the spin-coated films. In contrast, the variation in θ r is similar for the two types of surfaces, even if the change is more pronounced for the polymer networks.
The decrease in advancing water contact angle could be assigned to the change in dipole moment resulting from the trans to cis isomerization, in the same way as for azobenzene-modified polymers [16, 27, 30–32]. After irradiation, the polymer networks were thus heated in order to study the reversibility of the phenomenon. A heating for 3 h at 55 °C, i.e., the same temperature as for the solutions, was first carried out without any resulting contact angle variation. As mentioned previously, the reversibility was observed to be only partial in solution at such temperature. Considering the thermomechanical properties of the networks previously characterized by DMTA, the samples were then heated for 0.5 h at 130 °C, i.e., above the α-relaxation temperature of the networks. Indeed, one could thus expect higher polymer chain mobility. It was previously checked by means of thermogravimetry analysis that no polymer network degradation occurs under those conditions. AFM images obtained after sample irradiation and subsequent heating show no significant change in topography (ESM).
As shown in Fig. 10 for a CABg network corresponding to a number of cinnamate groups per cellulosic unit of 0.25, the advancing and receding contact angles go back to their initial values. Moreover, another cycle of photoirradiation heating can be performed with similar variations. The process is thus completely reversible whereas in solution, the cis to trans switching is partial upon heating at 55 °C. As reported by Hocking, it seems that the temperature has to be sufficiently high to allow the complete cis to trans switching [28]. In addition, it should be emphasized that the polymer networks are heated at a temperature exceeding the T α and thus promoting chain mobility. Even if the surface groups reorganize partially, the reversibility of the contact angle values during two cycles confirms that the dimerization is probably negligible. This could be related to the structure freezing through cross-linking of the cellulosic polymer with Desmodur® N3300 which leads to a high T α material. This probably prevents neighboring cinnamate groups to be in a configuration allowing their dimerization.

Variation of the advancing (a) and receding (b) contact angles measured on a CABg network (ds* = 0.25), as a function of time, for two cycles of photoirradiation (4 h)—heating (130 °C, 0 min)
Such reversible trans–cis process was extensively studied with azobenzene-containing polymer surfaces; however, to our knowledge, it was never reported for surfaces bearing cinnamate groups in which dimerization is usually searched for.
The difference in photochemical process according to the photosensitive polymer sample preparation has a marked influence on the contact angles values. Cross-linked cellulose derivatives bearing cinnamate groups are interesting candidates to obtain a reversible wetting modulation upon irradiation and heating. In contrast, spin-coating of cinnamate-modified cellulosic polymers leads to an irreversible dimerization process upon photoirradiation which is not compatible with reversible wetting modulation. This is probably related to the structure of the films which could be more disordered, allowing the photosensitive groups to be sufficiently close to each other to dimerize upon irradiation.
Conclusions
Cinnamate groups were grafted onto a cellulose derivative (CAB) with different densities according to the reaction time. Efficient grafting was demonstrated and quantified by SEC and 1H NMR analysis. The photochemistry of the cinnamate-modified CAB polymers was studied in solution by coupling UV–visible and 1H NMR spectroscopies. Upon UV irradiation, the trans to cis process occurs without any dimer formation while the switching can be only partly reversed by heating for 16 h at 55 °C.
The photochemical properties and wetting behavior of CAB-based spin-coated films and networks synthesized with an exogenous cross-linker were then characterized. The wetting properties of water on the networks obtained with different numbers of cinnamate groups per cellulosic unit were studied upon photoirradiation and then heating. The results indicate that the trans–cis isomerization is the main process at the surface. Indeed, photoirradiation leads to a decrease in advancing contact angle and an increase in receding contact angle, all the more since the density of cinnamate groups is high. Such variations are similar to those evidenced with azobenzene as photosensitive group and water as liquid. Moreover, the isomerization is completely reversible through heating at 130 °C and an extra cycle of photoirradiation heating can be performed leading to the same variations.
In contrast, dimerization is evidenced as the main process upon photoirradiation of the spin-coated films of cinnamate-modified CAB. This results in a significant difference in the wetting behavior of the surfaces with respect to the networks. In particular, dimerization prevents reversibility of contact angles upon heating of the irradiated surfaces.
Our study evidences the possible use of cinnamate as surface active groups, depending on the sample preparation, and further studies are in progress to emphasize surface roughness of the networks for amplification of light-induced contact angles switching.
References
Katsonis N, Lubomska M, Pollard MM, Feringa BL, Rudolf P (2007) Prog Surf Sci 82:407
Ichimura K, Oh SK, Nakagawa M (2000) Science 288:1624
Yang D, Piech M, Bell NS, Gust D, Vail S, Garcia AA, Schneider J, Park CD, Hayes MA, Picraux ST (2007) Langmuir 23:10864
Schannon PJ, Gibbons WM, Sun ST (1994) Nature 368:532
Wang S, Song Y, Jiang LJ (2007) Photochem Photobiol C: Photochem Rev 8:18
Xin B, Hao J (2010) Chem Soc Rev 39:769
Lim H, Han JT, Kwak D, Jin M, Cho K (2006) J Am Chem Soc 128:14458
Fang G, Li W, Wang X, Qiao G (2008) Langmuir 24:11651
Liu J-H, Chung Y-C, Lin M-T (1994) Die Angewandte Makromolekulare Chemie 219:43
Shibaev V, Bobrovsky A, Boiko N (2003) Prog Polym Sci 28:729
Lee SW, Kim SI, Lee B, Choi W, Chae B, Kim SB, Ree M (2003) Macromolecules 36:6527
Ichimura K, Akita Y, Akiyama H, Kudo K, Hayashi Y (1997) Macromolecules 30:903
Kong H, Lu X, Xiao S, Lu Q (2009) Polymer 50:1166
Gerus I, Glushchenko A, Kurioz Yu, Reznikov Yu, Tereshchenko O (2004) Opto-Electronics Review 12:281
Assaïd I, Hardy I, Bosc D (2002) Opt Commun 214:171
Feng C, Zhang Y, Jin J, Song Y, Xie L, Qu G, Jiang L, Zhu D (2001) Langmuir 17:4593
Paik MY, Krishnan S, You F, Li X, Hexemer A, Ando Y, Kang SH, Fischer DA, Kramer EJ, Ober CK (2007) Langmuir 23:5110
Jiang W, Wang G, He Y, Wang X, Song Y, Jiang L (2005) Chem Commun 28:3550
Gonçalves VC, Ferreira M, Olivati CA, Cardoso MR, Mendonça CR (2008) Colloid Polym Sci 286:1395
Delorme N, Bardeau J, Bulou A, Poncin-Epaillard F (2005) Langmuir 21:12278
Kamath M, Kincaid J, Mandal BK (1996) J Appl Polym Sci 59:45
Bouteau M, Cantin S, Fichet O, Perrot F, Teyssié D (2010) Langmuir 26:17427
Arai K, Satoh H (1992) J Appl Polym Sci 45:387
Laskar J, Vidal F, Fichet O, Gauthier C, Teyssie D (2004) Polymer 45:5047
Brandrup J, Immergut EJ, Grulke EA (1999) Polymer handbook, 4th edn. Wiley, New York
Ruslim C, Ichimura K (1999) Macromolecules 32:4254
Matsuhira T, Yamamoto H, Okamura T, Ueyama N (2008) Org Biomol Chem 6:1926
Aruna P, Rao BS (2009) React Funct Polym 69:20
Ito Y, Borecka B, Olovsson G, Trotter J, Scheffer JR (1995) Tetrahedron Lett 36:6087
Hocking MB (1969) Can J Chem 47:4567
Oh S, Nakagawa M, Ichimura K (2002) J Mater Chem 12:2262
Ding L, Russel TP (2007) Macromolecules 40:2267
Yuan W, Jiang G, Wang J, Wang G, Song Y, Jiang L (2006) Macromolecules 39:1300
Tylkowski B, Peris S, Giamberini M, Garcia-Valls R, Reina JA, Ronda JC (2010) Langmuir 26:14821
Sung SJ, Cho KY, Kim WS, Chang HS, Cho I, Park JK (2004) Chem Phys Lett 394:238
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
AFM images obtained on CABg spin-coated films and polymer networks are available in supporting information.
ESM 1
(DOCX 848 kb)
Rights and permissions
About this article
Cite this article
Abrakhi, S., Péralta, S., Cantin, S. et al. Synthesis and characterization of photosensitive cinnamate-modified cellulose acetate butyrate spin-coated or network derivatives. Colloid Polym Sci 290, 423–434 (2012). https://doi.org/10.1007/s00396-011-2559-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-011-2559-9