
Abstract
Inflammatory responses play an important role in the development of left ventricular (LV) hypertrophy and dysfunction. Recent studies demonstrated that increased T-cell infiltration and T-cell activation contribute to LV hypertrophy and dysfunction. Dendritic cells (DCs) are professional antigen-presenting cells that orchestrate immune responses, especially by modulating T-cell function. In this study, we investigated the role of bone marrow-derived CD11c+ DCs in transverse aortic constriction (TAC)-induced LV fibrosis and hypertrophy in mice. We observed that TAC increased the number of CD11c+ cells and the percentage of CD11c+ MHCII+ (major histocompatibility complex class II molecule positive) DCs in the LV, spleen and peripheral blood in mice. Using bone marrow chimeras and an inducible CD11c+ DC ablation model, we found that depletion of bone marrow-derived CD11c+ DCs significantly attenuated LV fibrosis and hypertrophy in mice exposed to 24 weeks of moderate TAC. CD11c+ DC ablation significantly reduced TAC-induced myocardial inflammation as indicated by reduced myocardial CD45+ cells, CD11b+ cells, CD8+ T cells and activated effector CD8+CD44+ T cells in LV tissues. Moreover, pulsing of autologous DCs with LV homogenates from TAC mice promoted T-cell proliferation. These data indicate that bone marrow-derived CD11c+ DCs play a maladaptive role in hemodynamic overload-induced cardiac inflammation, hypertrophy and fibrosis through the presentation of cardiac self-antigens to T cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systolic overload initiates cardiac hypertrophy and, eventually, congestive heart failure (CHF) [18]. In this process, pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β, are increased during CHF [3, 8, 18, 20]; these cytokines are known to induce cardiomyocyte hypertrophy and cardiac fibrosis following the inflammatory response [4]. Activation of Toll-like receptor 4 (TLR4) signaling pathway contributes to systolic overload-induced cardiac inflammation, fibrosis, and dysfunction [6]. Interestingly, recent studies demonstrated that T cells play an important role in promoting the pressure overload-induced left ventricular (LV) hypertrophy and dysfunction [16, 21]. Our recent study further demonstrated that chronic pressure overload causes an increase of activated effector T cells and inhibition of T-cell activation by B7 or CD28 gene deficiency can partially attenuate pressure overload-induced LV hypertrophy and dysfunction [23]. In addition, recent studies by us and others have consistently demonstrated that regulatory T cells (Tregs), a group of powerful anti-inflammatory T cells, can effectively suppress pressure overload-induced LV leukocyte and T-cell infiltration and LV hypertrophy in mice [12, 22], indicating an important role for T cells in regulating LV hypertrophy and dysfunction. However, the cellular and molecular mechanism of T-cell activation during LV hypertrophy and dysfunction is largely unknown.
Dendritic cells (DCs) are professional antigen-presenting cells that orchestrate immune responses in many pathological conditions by modulating T-cell activity [2]. Immature DCs have a high capacity to sense chemo-attracting inflammatory signals and mobilize to sites of infection or tissue destruction, where they engulf and process foreign materials or cell debris and present antigenic peptides on cell surface MHCII (major histocompatibility complex class II) receptors. These activated DCs then migrate to T-cell-rich areas such as the lymph node or spleen, where they stimulate the proliferation of antigen-specific T cells. Several recent studies demonstrate that endogenous antigen(s), particularly oxidized endogenous peptides, contribute to T-cell activation in Angiotensin-II-induced hypertension through modulating DCs in mice [13, 26]. However, it is not clear whether DCs also regulate T-cell activation in response to cardiac stress and tissue damage that occurs during pressure overload-induced LV hypertrophy.
In mice, CD11c is highly expressed on most DCs and some macrophages, two types of antigen-presenting cells. In the present study, we examined the role of CD11c+ cells in cardiac adaptation to LV pressure overload. We demonstrated that transverse aortic constriction (TAC)-induced LV hypertrophy is associated with accumulation of CD11c+ cells and CD11c+ MHCII+ cells in the LV, spleen and peripheral blood. Since CD11c+ MHCII+ cells are generally described as DCs, we will call the CD11c+ MHCII+ cells as CD11c+ MHCII+ DCs. Importantly, depletion of bone marrow (BM)-derived CD11c+ cells using a CD11c-DTR/GFP (CD11c-diphtheria toxin receptor/green fluorescent protein) transgenic mouse strain [1, 27] dramatically attenuated TAC-induced accumulation of CD45+ leukocytes, CD8+ T cells, and the CD8+ CD44+ T cell subset in the LV. To our surprise, depletion of BM-derived CD11c+ cells almost abolished TAC-induced LV fibrosis and hypertrophy. Furthermore, we demonstrated that depletion of BM-derived CD11c+ cells dramatically attenuated TAC-induced CD8+ T-cell proliferation. Collectively, our findings establish that BM-derived CD11c+ cells play an important role in pressure overload-induced T-cell activation, and LV fibrosis and hypertrophy, suggesting that therapies which target DC proliferation or activation may have potential for preventing cardiac hypertrophy and fibrosis.
Methods
Detailed methods are available in the online-only Data Supplement.
Animals and experiment design
CD11c-DTR/GFP mice (stock # 004509) and wild-type (WT) mice on a C57BL/6 background were purchased from Jackson Laboratory. Bone marrow cells (BMCs) from CD11c-DTR/GFP mice were transplanted into irradiated WT recipient mice (4 weeks old) [24]. Four weeks after transplantation of BMCs, these mice were subjected to moderate TAC using a 26G needle to create the aortic constriction [24]. Sham surgery was conducted on control mice. One day before TAC, some mice received intraperitoneally administered DT (Sigma) at a dose of 4 ng/g body weight every 3 days for the duration of the experiment to deplete CD11c+ DCs. PBS was injected as a control [1]. Final echocardiography was performed 24 weeks after TAC [24], and samples were collected for histological staining, quantitative real-time PCR and flow cytometric analysis. Primary antibodies for flow cytometric analysis used in this study are listed in Table S1. Studies were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
T-cell proliferation assay
The purpose of this assay was to determine whether CD11c+ cells pulsed by proteins from the LV of mice with LV hypertrophy after TAC could promote T-cell proliferation [26]. Briefly, the LV tissue was homogenized in PBS with Ca2+ and Mg2+ supplemented with protease inhibitor and phosphatase inhibitor phosSTOP (Roche). The protein concentration of the homogenate was measured using Bio-Rad Protein Assay. CD11c+ DCs were isolated from spleens; while pan T cells were isolated from mediastinal lymph nodes using corresponding magnetic cell sorting kits from Miltenyi Biotec. Pan T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) according to the manufacturer’s instructions. One million DCs from control mice were exposed to 100 μg LV proteins from control mice or mice with LV hypertrophy for 2 h, and then 0.5 × 105 DCs were co-cultured with 0.5 × 106 pan T cells from control mice or mice with LV hypertrophy for 4 days in a complete medium (RPMI 1640 with 10% FBS, 1% penicillin/streptomycin/fungizone, and 1 mM sodium pyruvate) in various combinations. These cells were analyzed by flow cytometry.
Statistics
Data are presented as mean ± SEM. A Student’s t test was used to test for differences between two groups. A two-way ANOVA followed by a Bonferroni correction post hoc test was used to test for differences among more than two groups. All pairwise P values are two-sided. The null hypothesis was rejected at P < 0.05.
Results
Chronic pressure overload causes accumulation and activation of DCs in LV tissues in mice
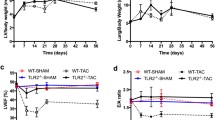
Our recent study demonstrated that T-cell accumulation and activation are increased in the LV tissues in mice after chronic TAC [23]. To determine whether LV inflammation was related to an increase of DCs, we examined LV CD11c+ cells and CD11c+ MHCII+ DCs under control conditions and 8 weeks after TAC in mice using flow cytometry analysis with the gating strategy are described in Suppl. Fig. 1. As shown in Fig. 1a, the ratio of LV weight to body weight were significantly increased ~2.0-fold in mice after chronic TAC. Echocardiography demonstrated a significant decrease of LV fractional shortening (FS) in mice after TAC (18.6% in TAC group vs 45.8% in control group) (Fig. 1b). Flow cytometry data showed that TAC caused significant increases of total CD11c+ cells and CD11c+ MHCII+ DCs in LV tissues (Fig. 1c, d), as well as the percentage of CD11c+MHCII+ DCs in CD11c+ cell subset and in CD45+ cell subset (Fig. 1e–h), indicating increased DC accumulation in LV tissues in mice after TAC. MHCII expressions in CD11c+ MHCII+ DCs and in CD11c+ cells were significantly increased in LV tissues from mice after TAC (Fig. 1i–k), further indicating an increased DC activation in these mice. These data demonstrate that TAC-induced systolic overload causes a significant increase of DC infiltration and activation in LV tissues.

Chronic transverse aortic constriction (TAC) results in recruitment of dendritic cells (DCs) in the left ventricle (LV) of mice. Data were collected from mice under control conditions (Ctr) and 8 weeks after TAC. a The ratio of LV weight to body weight of mice. b Echocardiographic measurements of LV fractional shortening (FS) of mice. c, d Quantitative data show total numbers of CD11c+ cells and CD11c+ MHCII+ (major histocompatibility complex class II molecules) cells in LV tissues. e–h Flow cytometry plots and quantitative data represent percentages of CD11c+ cells in CD45+ cells, CD11c+ MHCII+ cells in CD11c+ cells and CD11c+ MHCII+ cells in CD45+ cells in LV tissues. i–k, Flow cytometry histograms and quantitative data represent expression levels of MHCII on CD11c+ cells and CD11c+MHCII+ cells in LV tissues. n = 4–6 per group. *P < 0.05 vs control group
Chronic pressure overload increases and activates DCs in the spleen and peripheral blood in mice
Since our previous data showed that TAC caused a mild but significant increase of activated T cells in the spleen and peripheral blood in mice [23], we further examined DCs in spleen tissues and peripheral blood using flow cytometry analysis (gating strategies are presented in Suppl. Fig. 2). The percentages of CD11c+ MHCII+ DCs in total cells and in CD11c+ cell subset were markedly increased in spleen tissues from mice after chronic TAC (Fig. 2a, c, d). Additionally, the percentage of CD11c+ cells was significantly increased in spleen tissues and in peripheral blood from mice after chronic TAC (Fig. 2a, b, h, i). MHCII expressions in CD11c+ MHCII+ DCs and in CD11c+ cells were also significantly enhanced in spleen tissues from mice after chronic TAC (Fig. 2e–g). Collectively, these data indicate that TAC-induced systolic overload causes significant increases of DC accumulation and activation in spleen tissues and peripheral blood in mice.

Chronic transverse aortic constriction (TAC) results in recruitment of dendritic cells (DCs) in the spleen and peripheral blood of mice. Data were collected from mice under control conditions (Ctr) and 8 weeks after TAC. a–d Flow cytometry plots and quantitative data represent percentages of CD11c+ cells in splenocytes, and CD11c+ MHCII+ (major histocompatibility complex class II molecules) cells in CD11c+ cells and splenocytes. e–g Flow cytometry histograms and quantitative data show the relative expression level of MHCII in CD11c+ cells and CD11c+ MHCII+ cells in spleens. h, i Flow cytometry plots and quantitative data represent the percentage of CD11c+ cells in peripheral circulating blood cells. n = 4–6 per group. *P < 0.05 vs control group
Effective depletion of CD11c+ DCs in the LV of mice after TAC
Since increases of DC accumulation and activation in the LV, spleen and peripheral blood are associated with the development of LV hypertrophy and dysfunction, it is important to determine whether DCs contribute to the development of LV hypertrophy or dysfunction. It has been reported that DT injection into CD11c-DTR/GFP transgenic mice leads to rapid depletion of CD11c+ DCs [11]. DCs in the spleen were depleted more than 90% for the first 2 days after DT injection, and the number of DCs was gradually restored after 2 days [11]. However, a single DT injection leads to animal death in 6–7 days, whereas repeated injection of DT into WT mice reconstituted with BMCs from CD11c-DTR/GFP mice could effectively deplete BM-derived DCs with no apparent deleterious effects [27]. Therefore, we transplanted BMCs from CD11c-DTR/GFP mice into irradiated WT recipients. Moderate TAC procedure was then performed in these mice 4 weeks after the transplantation of BMCs. One day before TAC, some mice began to receive DT treatment at a dose of 4 ng/g body weight i.p. every 3 days to deplete DCs. Cardiac function and tissue sampling were performed 24 weeks after TAC (Fig. 3a). In CD11c-DTR/GFP-BMC chimeric mice, we found that spleen CD11c+ cells were depleted more than 90% 2 days after the first DT injection (Suppl. Fig. 3).

Strategy of depletion of bone marrow-derived dendritic cells (DCs) in mice. a Diagram of experimental design. One day before chronic transverse aortic constriction (TAC), chimeric mice reconstituted with bone marrow cells from CD11c-DTR/GFP (CD11c-diphtheria toxin receptor/green fluorescent protein transgenic) mice were administrated diphtheria toxin to deplete DCs in vivo. Data were collected from chimeric mice under control conditions (Ctr), or treated with PBS or DT under TAC conditions (TAC or DC-ablation TAC). b–d Flow cytometry plots and quantitative data represent percentages of GFP+ cells, GFP+ MHCII+ (major histocompatibility complex class II molecules) cells in CD45+ cells from LV tissues. e–g Flow cytometry plots and quantitative data represent percentages of CD11c+ cells, CD11c+ MHCII+ cells in CD45+ cells from LV tissues. n = 4–6 per group. *P < 0.05 vs control group; # P < 0.05 vs TAC group treated with PBS
Flow cytometry analysis of LV CD45+ cells revealed that most of GFP+ cells or CD11c+ cells in the LV after TAC were MHCII positive (Fig. 3b, e). The percentages of GFP+MHCII+ DCs and GFP+ cells were increased in the LV after TAC (Fig. 3b–d). DT administration depleted ~80% of GFP+MHCII+ DCs and GFP+ cells after TAC (Fig. 3b–d). This observation was consistent with the result from CD11c+MHCII+ DCs (Fig. 3e–g). These data demonstrate that DT treatment of CD11c-DTR/GFP-BMC chimeric mice effectively depletes CD11c+ DCs in the LV after TAC.
Depletion of CD11c+ DCs attenuates TAC-induced LV hypertrophy and fibrosis in mice
Under control conditions, DC ablation had no detectable effect on the ratio of LV weight to body weight, myocyte size or LV fibrosis in CD11c-DTR/GFP-BMC chimeric mice (Figs. 4a–c, 5a–c). Echocardiographic measurements showed that DC ablation did not affect LV FS and LV dimensions under control conditions (Fig. 5e–g). These data indicate that DC ablation with DT treatment has no detectable effect on cardiac structure or function under control conditions.

Depletion of bone marrow-derived dendritic cells (DCs) attenuates chronic transverse aortic constriction (TAC)-induced left ventricular (LV) hypertrophy in mice. Data were collected from chimeric mice under control conditions (Ctr), or treated with PBS or diphtheria toxin after TAC (TAC or DC-ablation TAC). a, b Representative images and quantitative data show the LV myocyte size of mice, as determined by FITC-conjugated wheat germ agglutinin (WGA) staining. c The ratio of LV weight to body weight of mice. *P < 0.05 vs control group; # P < 0.05 vs TAC group of WT mice

Depletion of bone marrow-derived dendritic cells (DCs) attenuates chronic transverse aortic constriction (TAC)-induced left ventricular (LV) fibrosis and inflammation in mice. Data were collected from chimeric mice under control conditions (Ctr), or treated with PBS or diphtheria toxin after TAC (TAC or DC-ablation TAC). a, b Representative images and quantitative data of Sirius red/Fast green staining for detection of fibrosis in the LV. c, d Quantitative RT-PCR results of TGF-β and IL-1β mRNA levels in LV lysates. e–g Echocardiographic measurements of LV fractional shortening (FS), LV end-systolic diameter (LVESD) and LV end-diastolic diameter (LVEDD) of mice. *P < 0.05 vs control group; # P < 0.05 vs TAC group of WT mice
TAC caused significant LV hypertrophy 1.5-fold in CD11c-DTR/GFP-BMC chimeric mice (Fig. 4a–c; Table S2). Examination of the ratio of LV weight to body weight and myocyte size revealed that DC ablation remarkably attenuated the TAC-induced increase in LV hypertrophy (Fig. 4a–c; Table S2). As shown in Fig. 5a, b, LV fibrosis as identified by Sirius red and Fast green staining was remarkably increased (5.1-fold) in mice after TAC as compared to control mice. DC ablation significantly attenuated the TAC-induced LV fibrosis. In addition, TAC-induced increases of LV mRNA contents of TGF-β (a pro-fibrogenic cytokine) and IL-1β (a pro-inflammatory cytokine) were significantly attenuated by DC ablation (Fig. 5c, d). TAC did not affect mRNA expressions of TNF-α and IL-10 in the LV (data not shown). These data demonstrate that DC ablation attenuates systolic overload-induced LV fibrosis and inflammation in mice. Echocardiographic measurements showed that depletion of DCs significantly attenuated the TAC-induced reduction of LV FS and the TAC-induced increase of LV end-systolic diameter (LVESD) in mice, although TAC did not induce obvious LV dysfunction in both groups (FS: 39.0% in TAC group vs 45.7% in control group) (Fig. 5e–g).
Depletion of CD11c+ DCs attenuates TAC-induced leukocyte infiltration in the LV of mice
LV hypertrophy development is associated with inflammation and immune responses that are orchestrated by CD11c+ DCs. In order to understand the mechanism of the beneficial effects of DC ablation on the LV inflammation due to TAC, we examined total CD45+ cells, CD11b+ cells, CD8+ T cells and activated effector CD8+ T cells (CD8+CD44+ cells) in the LV using a flow cytometry analysis. TAC caused a significant accumulation of CD45+ cells (2.2-fold), CD11b+ cells (1.7-fold), CD8+ T cells (3.8-fold) and CD8+CD44+ activated effector T cells (8.0-fold) in the LV of PBS-treated CD11c-DTR/GFP-BMC chimeric mice. Remarkably, DC ablation with DT treatment significantly reduced all TAC-induced leukocyte accumulations in the LV of CD11c-DTR/GFP-BMC chimeric mice (Fig. 6a–n).

Depletion of bone marrow-derived dendritic cells (DCs) decreases chronic transverse aortic constriction (TAC)-induced leukocyte infiltration in the left ventricle (LV) of mice. Data were collected from chimeric mice under control conditions (Ctr), or treated with PBS or diphtheria toxin after TAC (TAC or DC-ablation TAC). a, b Representative images and quantitative data of CD45 immunostaining (red) of the LV. c–n Flow cytometry plots and quantitative data represent percentages of CD45+ cells, CD11b+ cells, CD8+ cells and activated effector CD8+ cells (CD8+CD44+ cells) in the LV. n = 4–6 per group. *P < 0.05 vs control group; # P < 0.05 vs TAC group treated with PBS
As expected, DT administration in the CD11c-DTR/GFP-BMC chimeric mice depleted 99% of CD11c+ MHCII+ cells in the spleen (Suppl. Fig. 4a). Moreover, DC ablation also led to a significant reduction of the percentage of CD8+ T cells in the spleen (Suppl. Fig. 4b). Collectively, these data suggest that DC depletion effectively attenuates systolic overload-induced immune-cell accumulation in the LV and spleen of mice.
DCs pulsed with cardiac proteins from mice with LV hypertrophy promote T-cell proliferation
CD11c+ DCs initiate the immune response by presenting their antigenic peptide/MHCII complexes to T cells. To determine whether TAC-produced cardiac proteins act as antigenic peptides for DCs to stimulate autoreactive T cells, we used LV homogenates from control or LV-hypertrophy mice to pulse naïve CD11c+ DCs from spleens of normal WT mice. These pulsed DCs were then co-cultured with CFSE-labeled pan T cells isolated from mediastinal lymph nodes of control mice or LV-hypertrophy mice (Fig. 7a). Interestingly, flow cytometry showed that DCs pulsed with the LV homogenate from LV-hypertrophy mice promoted the proliferation of both CD4+ and CD8+ T cells isolated from LV-hypertrophy mice; while DCs pulsed with the control LV homogenate had no effect on the proliferation of CD4+ or CD8+ T cells isolated from either normal or LV-hypertrophy mice. Moreover, DCs pulsed with the LV homogenate from LV-hypertrophy mice also had no detectable effect on the proliferation of CD4+ or CD8+ T from control mice (Fig. 7b, Suppl. Fig. 5). These data show that DCs provoked by antigenic peptides in the LV of LV hypertrophy mice can prime T cells in draining lymph nodes, suggesting that immunogenic DCs promote LV inflammation in LV hypertrophy mice.

Dendritic cells (DCs) pulsed with cardiac proteins from mice with left ventricular (LV) hypertrophy promote T-cell proliferation. a Diagram of experimental design. CD11c+ cells pulsed with LV homogenates from normal control mice (Ctr) or mice with LV hypertrophy after chronic transverse aortic constriction (TAC) were co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled pan T cells isolated from the mediastinal lymph nodes of control or mice with LV hypertrophy in various combinations. b Flow cytometry histograms represent percentages of proliferating CD4+ and CD8+ T cells, as determined by CSFE dilution. n = 3 per group. *P < 0.05 vs control group
Discussion
In this study, we show that chronic LV pressure overload leads to significant increases in the number of CD11c+ cells in the heart, spleen and peripheral blood. Chronic pressure overload also significantly increases the DC activation as evidenced by the significant increase of MHCII expressions in CD11c+ MHCII+ cells and in CD11c+ cells. In addition, we provide evidence that BM-derived CD11c+ DCs contribute to the pathological LV remodeling in response to pressure overload. Specifically, by using CD11c-DTR/GFP-BMC chimeric mice, we demonstrated that ablation of CD11c+ DCs prevents TAC-induced LV fibrosis and hypertrophy, ostensibly by limiting myocardial DC antigenic peptide presentation for T-cell proliferation, as well as subsequent inflammatory cell infiltration in LV tissues. Moreover, we demonstrated that depletion of BM-derived CD11c+ DCs significantly attenuates TAC-induced increases of cardiac CD8+ CD44+ T cells, suggesting an important role of CD11c+ DCs in regulating TAC-induced T-cell activation. Furthermore, we demonstrated that DCs stimulated ex vivo by myocardial proteins from LV-hypertrophy mice promoted the proliferation of T cells isolated from heart-draining lymph nodes of LV-hypertrophy mice. Collectively, these findings indicate that DC accumulation and activation in the LV are maladaptive in the setting of LV pressure overload, suggesting that strategic targeting of DC infiltration or activation may be an efficacious therapeutic approach to ameliorate pressure overload-induced LV fibrosis and hypertrophy.
DCs play a critical role in orchestrating immune responses to various pathological insults—with both positive and negative effects. Using a mouse myocardial infarction model, a recent study revealed that DC depletion exacerbated LV dysfunction and remodeling after coronary artery ligature by enhancing inflammatory monocyte/macrophage recruitment to the LV [1]. Data shown here now provide evidence that chronic pressure overload induces the accumulation and activation of DCs within the LV, spleen, and peripheral blood, and these DCs then promote cardiac inflammation and hypertrophy in mice. The striking reduction in TAC-induced LV inflammation and hypertrophy in mice depleted of BM-derived CD11c+ DCs indicates that DCs are one of major maladaptive factors in pressure overload-induced cardiac inflammation and hypertrophy. Markedly reduced accumulation of CD45+ cells, including CD11b+ cells and activated effector CD8+ T cells in the LV of CD11c+ cell-depleted mice suggests that DCs orchestrate a cellular immune response that targets ventricular tissue, which promotes myocardial inflammation. Our finding that DCs pulsed with an LV homogenate from LV-hypertrophy mice can induce the proliferation of mediastinal CD4+ and CD8+ T cells from LV-hypertrophy mice indicates that a DC-dependent breakdown in T-cell tolerance to a myocardial self-antigen(s) and clonal expansion underlies this process.
Increasing evidence shows that immune cells are critically involved in maladaptive hypertrophic response, which leads to the transition from LV hypertrophy to heart failure [5]. Studies demonstrate an important role for T cells, effector memory T cells, and Tregs in the development of LV hypertrophy and dysfunction. One study showed that depletion of T cells in either Rag2 or CD4 knockout mice led to an attenuation of myocardial inflammation and fibrosis after TAC, and inhibited the transition from LV hypertrophy to LV dysfunction [16]. Recently, we demonstrated that activated T cells were accumulated in LV tissues from mice after TAC, and inhibition of T-cell co-stimulatory signaling using CD28 or B7 knockout mice effectively attenuated TAC-induced T-cell activation, and LV hypertrophy and dysfunction [23]. Moreover, the induction of endogenous anti-inflammatory Tregs with interleukin-2 (IL-2) plus IL-2 antibody complexes significantly attenuated TAC-induced LV hypertrophy and dysfunction, as well as the progression from LV failure to RV hypertrophy [22]. These results are compatible with the observation that an adoptive transfer of autologous Tregs can reduce pressure overload-induced cardiac inflammation and hypertrophy [12, 15]. Taken together, the findings indicate an important role of acquired immunity in the development of LV hypertrophy and dysfunction [5]. Since CD11c+ DCs play an important role in T-cell proliferation and activation, the increased LV DC infiltration, particularly activated DCs induced by a stressed and overloaded myocardium, is anticipated to cause LV inflammation, fibrosis and hypertrophy in mice at least in part through self-antigen-specific stimulation of T-cell activation and proliferation.
Antigen-presenting cells, including macrophages, DCs, Langerhans cells, and B-lymphocytes mediate cellular immune response by processing and presenting antigens to the T-cell receptor. The role of macrophages in cardiac inflammation and remodeling has been widely studied. It has been reported that depletion of macrophages using clodronate liposomes in hypertensive rats accelerates cardiac dysfunction without affecting cardiac hypertrophy, suggesting that macrophages are important for cardiac repair [28]. Inhibition of monocyte trafficking using CCR2 or a monocyte chemoattractant protein-1 knockout or their neutralizing antibodies significantly reduced pressure overload-induced macrophage infiltration and myocardial fibrosis, without affecting cardiac hypertrophy [7, 10, 14, 19]. Inhibition of monocyte recruitment using a CCR2 inhibitor attenuated myocardial inflammation and injury repair due to a DT-induced cardiomyocyte death and apoptosis in mice [17]. Moreover, manipulation of macrophage polarization reduced cardiac hypertrophy and fibrosis, and preserved cardiac function [9]. We recently demonstrated that inhibition of monocyte trafficking using genetic deletion of CCR2 or pharmacological inhibition led to attenuated Ang-II or DOCA salt-induced vascular inflammation, fibrosis and vasodilation [25]. These studies demonstrate that macrophages also play either a deleterious role or a beneficial role in cardiac injury repair and pressure overload-induced LV hypertrophy and dysfunction. Our present data provide direct evidence for the first time that DCs promote pressure overload-induced LV inflammation, fibrosis and hypertrophy.
The present study has several limitations. First, since the current experimental approach could only effectively deplete the BM-derived CD11c+ DCs without depleting residential CD11c+ DCs (Suppl. Fig. 6), the reduced LV inflammation, fibrosis and hypertrophy observed in our study should only be interpreted as the effect of BM-derived CD11c+ DCs. The overall impact of CD11c+ DCs on the development of LV fibrosis and hypertrophy might have been underestimated by this experimental approach. In addition, since CD11c is also expressed on some activated macrophages, the experimental approach used in the present study might also deplete CD11c+ macrophages. Moreover, our study could not differentiate relative role of depletion of CD11c+ DCs and CD11c+ macrophages in attenuating TAC-induced LV fibrosis and hypertrophy. As some DCs does not express CD11c, the pathological effect of CD11c− DCs in the pressure overload-induced LV fibrosis and hypertrophy could not be addressed in the present study. In addition, in order to observe the effect of chronic pressure overload on LV hypertrophy, and to reduce unwanted postsurgical mortality rate, we used a moderate TAC procedure in the chimeric mice. While the moderate TAC procedure caused 1.5-fold LV hypertrophy; unfortunately, these mice did not develop severe LV dysfunction even after 6 months of moderate TAC. Therefore, our data could only address the role of BM-derived DCs in regulating LV hypertrophy and fibrosis. Moreover, as human DCs and mouse DCs express different cell surface markers, the findings observed in mouse studies might be different to the clinical conditions. Nevertheless, our findings provide the first direct evidence of BM-derived DCs in regulating pressure overload-induced cardiac inflammation and the development of LV hypertrophy and fibrosis.
Perspectives
Left ventricular (LV) hypertrophy and dysfunction are accompanied by increased cardiac leukocyte infiltration. CD11c+ dendritic cells (DCs) are the major antigen-presenting cells that orchestrate systemic and tissue immune responses under various pathological conditions. The DC number and activation are significantly increased in the mice with LV hypertrophy. Using chimeric mice reconstituted with bone marrow cells from CD11c-DTR/GFP mice to selectively deplete CD11c+ DCs, we demonstrate that CD11c+ DCs promote systolic overload-induced LV inflammation, fibrosis and hypertrophy. Our study provides the first direct evidence that inhibiting bone marrow-derived CD11c+ DC proliferation or activation may be a new therapeutic approach for treating LV fibrosis and hypertrophy.
References
Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K (2012) Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125:1234–1245. doi:10.1161/CIRCULATIONAHA.111.052126
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252. doi:10.1038/32588
Diwan A, Tran T, Misra A, Mann DL (2003) Inflammatory mediators and the failing heart: a translational approach. Curr Mol Med 3:161–182
El-Menyar AA (2008) Cytokines and myocardial dysfunction: state of the art. J Card Fail 14:61–74. doi:10.1016/j.cardfail.2007.09.006
Frieler RA, Mortensen RM (2015) Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131:1019–1030. doi:10.1161/CIRCULATIONAHA.114.008788
Han J, Zou C, Mei L, Zhang Y, Qian Y, You S, Pan Y, Xu Z, Bai B, Huang W, Liang G (2017) MD2 mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF-kappaB signaling pathway. Basic Res Cardiol 112:9. doi:10.1007/s00395-016-0599-5
Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML (2010) Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 49:499–507. doi:10.1016/j.yjmcc.2010.05.005
Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K, Isoda K, Riches DW, Matoba S, Okigaki M, Matsubara H (2009) Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res 105:1149–1158. doi:10.1161/CIRCRESAHA.109.208199
Ikeda J, Ichiki T, Matsuura H, Inoue E, Kishimoto J, Watanabe A, Sankoda C, Kitamoto S, Tokunou T, Takeda K, Fong GH, Sunagawa K (2013) Deletion of phd2 in myeloid lineage attenuates hypertensive cardiovascular remodeling. J Am Heart Assoc 2:e000178. doi:10.1161/JAHA.113.000178
Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, Egashira K (2004) Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res 94:1203–1210. doi:10.1161/01.RES.0000126924.23467.A3
Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA (2002) In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17:211–220
Kanellakis P, Dinh TN, Agrotis A, Bobik A (2011) CD4(+)CD25(+)Foxp3(+) regulatory T cells suppress cardiac fibrosis in the hypertensive heart. J Hypertens 29:1820–1828. doi:10.1097/HJH.0b013e328349c62d
Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG (2014) DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 124:4642–4656. doi:10.1172/JCI74084
Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T (2004) Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension 43:739–745. doi:10.1161/01.HYP.0000118584.33350.7d
Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN (2009) Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119:2904–2912. doi:10.1161/CIRCULATIONAHA.108.832782
Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N (2014) CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129:2111–2124. doi:10.1161/CIRCULATIONAHA.113.007101
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL (2014) Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA 111:16029–16034. doi:10.1073/pnas.1406508111
Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK (1996) The progression from hypertension to congestive heart failure. JAMA 275:1557–1562
Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA (2008) Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 52:256–263. doi:10.1161/HYPERTENSIONAHA.108.112706
Mann DL (2015) Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116:1254–1268. doi:10.1161/CIRCRESAHA.116.302317
Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P (2015) Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail 8:776–787. doi:10.1161/CIRCHEARTFAILURE.115.002225
Wang H, Hou L, Kwak D, Fassett J, Xu X, Chen A, Chen W, Blazar BR, Xu Y, Hall JL, Ge JB, Bache RJ, Chen Y (2016) Increasing regulatory T cells with interleukin-2 and interleukin-2 antibody complexes attenuates lung inflammation and heart failure progression. Hypertension 68:114–122. doi:10.1161/HYPERTENSIONAHA.116.07084
Wang H, Kwak D, Fassett J, Hou L, Xu X, Burbach BJ, Thenappan T, Xu Y, Ge JB, Shimizu Y, Bache RJ, Chen Y (2016) CD28/B7 deficiency attenuates systolic overload-induced congestive heart failure, myocardial and pulmonary inflammation, and activated t cell accumulation in the heart and lungs. Hypertension 68:688–696. doi:10.1161/HYPERTENSIONAHA.116.07579
Wang H, Xu X, Fassett J, Kwak D, Liu X, Hu X, Falls TJ, Bell JC, Li H, Bitterman P, Bache RJ, Chen Y (2014) Double-stranded RNA-dependent protein kinase deficiency protects the heart from systolic overload-induced congestive heart failure. Circulation 129:1397–1406. doi:10.1161/CIRCULATIONAHA.113.002209
Wang L, Zhao XC, Cui W, Ma YQ, Ren HL, Zhou X, Fassett J, Yang YZ, Chen Y, Xia YL, Du J, Li HH (2016) Genetic and pharmacologic inhibition of the chemokine receptor CXCR2 prevents experimental hypertension and vascular dysfunction. Circulation 134:1353–1368. doi:10.1161/CIRCULATIONAHA.115.020754
Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ 2nd, Madhur MS, Harrison DG (2016) Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest 126:50–67. doi:10.1172/JCI80761
Zammit DJ, Cauley LS, Pham QM, Lefrancois L (2005) Dendritic cells maximize the memory CD8 T cell response to infection. Immunity 22:561–570. doi:10.1016/j.immuni.2005.03.005
Zandbergen HR, Sharma UC, Gupta S, Verjans JW, van den Borne S, Pokharel S, van Brakel T, Duijvestijn A, van Rooijen N, Maessen JG, Reutelingsperger C, Pinto YM, Narula J, Hofstra L (2009) Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. J Cardiovasc Pharmacol Ther 14:68–75. doi:10.1177/1074248408329860
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Sources of funding
This study was supported by Grants R01HL105406 and T32HL069764 from the National Institutes of Health, research Grants 81470512 and 81570355 from National Natural Science Foundation, and Research Grant 09GRNT2260175 from the American Heart Association.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, H., Kwak, D., Fassett, J. et al. Role of bone marrow-derived CD11c+ dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res Cardiol 112, 25 (2017). https://doi.org/10.1007/s00395-017-0615-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-017-0615-4