
Abstract
Accelerated development of coronary atherosclerosis is a defining characteristic of familial hypercholesterolemia (FH). However, the recent data highlight a significant cardiovascular risk prior to the development of critical coronary stenosis. We, therefore, examined the hypothesis that FH produces coronary microvascular dysfunction and impairs coronary vascular control at rest and during exercise in a swine model of FH. Coronary vascular responses to drug infusions and exercise were examined in chronically instrumented control and FH swine. FH swine exhibited ~tenfold elevation of plasma cholesterol and diffuse coronary atherosclerosis (20–60 % plaque burden). Similar to our recent findings in the systemic vasculature in FH swine, coronary smooth muscle nitric oxide sensitivity was increased in vivo and in vitro with maintained endothelium-dependent vasodilation in vivo in FH. At rest and during exercise, FH swine exhibited increased myocardial O2 extraction resulting in reduced coronary venous SO2 and PO2 versus control. During exercise in FH swine, the transmural distribution of coronary blood flow was unchanged; however, a shift toward anaerobic cardiac metabolism was revealed by increased coronary arteriovenous H+ concentration gradient. This shift was associated with a worsening of cardiac efficiency (relationship between cardiac work and O2 consumption) in FH during exercise owing, in part, to a generalized reduction in stroke volume which was associated with increased left atrial pressure in FH. Our data highlight a critical role for coronary microvascular dysfunction as a contributor to impaired myocardial O2 balance, cardiac ischemia, and impaired cardiac function prior to the development of critical coronary stenosis in FH.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Familial hypercholesterolemia (FH) is the most common monogenic disorder of lipoprotein metabolism resulting in pronounced lifetime hypercholesterolemia with plasma low-density lipoprotein (LDL) levels ranging between 190 and 1100 mg/dl [31]. Recent reports have nearly doubled prior estimates of FH prevalence to 1 in 200–300 individuals worldwide and have drawn attention to the underdiagnosis and undertreatment of this disorder, still considered by many clinicians to be rare [9, 21, 40]. FH is associated with markedly increased cardiovascular risk, particularly due to accelerated coronary atherosclerosis [31, 40]. A thorough understanding of the mechanistic basis of coronary mortality associated with FH, however, is lacking.
Accumulating evidence recognizes coronary microvascular dysfunction as an important factor contributing to cardiovascular mortality associated with co-morbid conditions, such as hypercholesterolemia and atherosclerosis [13, 41, 43, 52]. Recent clinical studies highlight the relationship of coronary microvascular dysfunction to poor outcomes during the progression of coronary atherosclerosis in patients with “intermediate” (40–70 % luminal narrowing) coronary artery stenosis [52]. Specifically, impaired coronary flow velocity reserve, an index of microvascular function, is associated with a significantly increased major adverse cardiovascular event rate in patients with “intermediate” coronary artery stenosis [52]. Similarly, reduced coronary flow reserve has been reported in patients with FH in the absence of flow-limiting coronary stenosis [30, 38, 54]. Indeed, a central role for coronary microvascular dysfunction owing to co-morbidity-associated systemic inflammation has recently been proposed to underlie the development of cardiac diastolic dysfunction [41]. Animals and humans with hypercholesterolemia demonstrate impaired coronary arteriolar dilation to pharmacologic agents in vivo [10, 38, 55] and ex vivo [25, 29, 46, 48, 53] that precede atherosclerotic plaque development [10, 55]. Taken together, these data challenge the stenosis-centered paradigm of cardiac ischemia in hypercholesterolemia and FH and highlight a need for a better understanding of coronary vascular function and blood flow control prior to the development of a critical coronary stenosis.
We recently reported, in chronically instrumented animals, impaired systemic and pulmonary vascular function in vivo during exercise in the Rapacz swine model of FH owing to endothelial dysfunction [2, 8]. These swine recapitulate human FH and its consequences (i.e., coronary atherosclerosis) due to reduced LDL catabolism and defective receptor binding of buoyant LDL [7, 32, 42, 44]. Given the lack of information regarding in vivo coronary blood flow control in response to physiologically relevant stressors, we utilized this model to evaluate coronary flow control in the presence of non-flow-limiting stenosis during exercise in FH. Specifically, we tested the hypothesis that FH produces coronary microvascular endothelial dysfunction and impairs coronary vascular control at rest and particularly during exercise compared to normal swine.
Methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Missouri and thus were performed in accordance with the Declaration of Helsinki (1964) and its later amendments. Adult (1–2 years) control, normolipidemic (Con, n = 7; 25–60 kg), or Rapacz familial hypercholesterolemic (FH, n = 4; 80–100 kg) swine were utilized. FH swine were obtained from the University of Wisconsin Swine Research and Teaching Center. Control swine were fed standard chow (by weight 16.7 % protein, 2.6 % fat, 53.2 % carbohydrate; 2.57 kcal/g). FH swine were fed standard chow until 14 mo of age when they were switched to a high-fat chow (by weight 13 % protein, 21.3 % fat, and 41.4 % carbohydrate; 5.14 kcal/kg) for 5 mo prior to instrumentation. Daily caloric intake was ~51 kcal/kg for control swine and ~48 kcal/kg for FH swine. FH swine were maintained on high-fat chow following instrumentation and throughout experimental protocols.
Instrumentation
Daily adaptation of animals to laboratory conditions and treadmill running began 1 week prior to surgery. Animals were sedated with telazol (5 mg/kg, im) and xylazine (2.2 mg/kg, im), intubated and ventilated with 1–3 % (vol/vol) isoflurane in the air to induce and maintain anesthesia. Body temperature was maintained between 36.5 and 37.5 °C using a warming blanket, and ECGs were monitored from the standard limb leads. Under sterile conditions, the chest was opened via the third intercostal space, and a fluid-filled polyvinylchloride catheter was inserted into the aortic arch for mean arterial pressure (MAP) measurement, blood sampling for PO2, PCO2, pH, O2 saturation, and hemoglobin concentration, the computation of O2 content, O2 supply, and O2 consumption [15, 34], as well as the collection of reference samples for microsphere studies [14]. A fluid-filled catheter was also inserted into the left atrium for measurement of the left atrial pressure (LAP) and infusion of microspheres and in the pulmonary artery for the blood sampling and infusion of drugs. A small angiocatheter was inserted into the anterior interventricular vein for coronary venous blood sampling. Finally, bidirectional transit-time flow probes (Transonic Systems) were placed around either the ascending aorta or pulmonary artery for the measurement of cardiac output and around the proximal left anterior descending coronary artery for the measurement of coronary blood flow (CBF).
Electrical wires and catheters were tunneled subcutaneously to the back, the chest was closed, and animals were allowed to recover. Animals received analgesia [buprenorphine (0.3 mg im) for 2 days] and antibiotic prophylaxis [amoxicillin (25 mg/kg iv) and gentamycin (5 mg/kg iv) for 5 days]. Catheters were flushed daily with physiological saline containing 1500–5000 IU/ml heparin. Studies were performed starting approximately 1 week after surgery, similar to the previous studies [2, 4].
Infusion protocols
The effect of FH on coronary endothelium-dependent and -independent vasodilation was assessed via infusion of the endothelium-dependent vasodilator ATP, the nitric oxide (NO) donor sodium nitroprusside (SNP), and the ATP-sensitive K+ (KATP) channel opener bimakalim, similar to the previous studies [8, 14, 35]. With swine standing quietly on the treadmill, resting hemodynamic measurements were collected with the subsequent infusion of agonists. Graded infusions of ATP (100–300 µg kg−1 min−1 iv), SNP (2–4 µg kg−1 min−1 iv), and bimakalim (75–225 ng kg−1 min−1 iv) were performed with each dose infused for 10 min and stable hemodynamics recorded after 6–8 min of each dose.
Exercise protocol
The effect of FH on CBF control at rest and during exercise was assessed via a treadmill exercise protocol. Briefly, with swine standing quietly on the treadmill, resting hemodynamic measurements, blood samples, and rectal temperature were obtained. Subsequently, control swine were subjected to a four-stage treadmill exercise protocol [2–5 miles/h (mph) at 0 % inclination], and FH swine were subjected to a three-stage exercise protocol [1–3 mph at 0 % inclination]. FH swine, in general, would not exercise beyond 3 mph. Hemodynamic variables were continuously recorded digitally on a Codas workstation (ATCODAS, Dataq Instruments, Akron, OH) with blood samples collected during the final 30 s of each 3-min exercise stage when steady-state hemodynamics had been achieved. Blood samples were analyzed for PO2, PCO2, pH, O2 saturation, and hemoglobin concentration (ABL-720, Radiometer, Copenhagen).
Myocardial blood flow
The transmural distribution of myocardial blood flow was measured by the injection of 6–8 × 106 microspheres [15 µm in diameter labeled with lanthanum, iridium, europium, holmium, ytterbium, samarium, scandium, lutetium, or terbium stable isotopes (BioPAL, Worcester, MA)] into the left atrium at rest and during exercise at 3 mph in the majority of animals. Injection was performed at 4 mph in five control swine. An arterial blood reference sample was withdrawn at a constant rate of 5 ml/min starting 10 s before the injection of microspheres and continuing for 90 s. At the conclusion of the study, swine were euthanized and the heart was excised and weighed. The left ventricular anterior wall was separated from the heart and divided into four layers of equal thickness from epicardium to endocardium. Samples from each layer were weighed and dried in individual vials along with reference blood samples. Myocardial and reference blood samples were sent to BioPAL for the analysis of microsphere content via neutron activation technology [45]. Blood flow per gram of myocardium (Q m) was calculated as: Q m = Q r × D m/D r, where Q r is the rate of withdrawal of the reference blood sample (in ml/min), D m is the disintegrations per minute per gram of the myocardial specimen, and D r is the disintegrations per minute of the reference blood sample.
Intravascular ultrasound (IVUS)
Coronary atherosclerosis was assessed by IVUS prior to sacrifice in FH swine. IVUS was initiated using the standard coronary catheterization techniques as described previously [17, 18, 28, 50, 51]. Briefly, a 7F introducer was inserted in the right femoral artery. Heparin (300 U/kg) was administered following femoral access and maintenance doses given every hour (100 U/kg). A guide catheter (6F) was directed up the aorta and engaged into the left ostium under fluoroscopic guidance. The left anterior descending (LAD) artery was selectively engaged. IVUS pullbacks (0.5 mm/s; Galazy II, Boston Scientific, 40 MHz) were obtained for the proximal, mid, and distal 3 cm segments of the LAD after intracoronary nitroglycerin. The 3D reconstruction of IVUS pullbacks was performed using the QIVUS software (Medis). Percent plaque (i.e., percent vessel volume reduction) was calculated as (plaque volume/vessel volume) × 100 in the proximal, mid, and distal segments.
Isolated arterioles
Apical left ventricular coronary arterioles from control and FH swine (<150 µm internal diameter) were isolated, mounted on glass micropipettes, and pressurized to 60 cmH2O for determination of vasodilator responses, as previously described [3]. Following 1 h equilibration, arterioles were preconstricted with endothelin-1. Endothelium-dependent and –independent vasodilator responses to bradykinin (BK; 3 fM–10 nM) and sodium nitroprusside (SNP; 1 nM–100 µM), respectively, were determined. Maximal passive diameter was determined at the end of the experiment by replacing the vessel bath with Ca2+-free buffer.
Data acquisition and analysis
All signals were recorded at a sampling rate of 225 Hz, digitized online, and stored on a computer for later post acquisition offline analysis with a program written in MatLab (MathWorks, Natick, MA). Mean values for in vivo measurements were determined across 10 s at rest and at the end of each exercise level. Systemic and coronary hemodynamic measurements as well as blood gas variables were analyzed, as previously described [15]. MVO2 was calculated as the product of CBF (normalized per gram of myocardium via blood flows determined by microspheres; CBF g−1) and the difference in O2 content between arterial and coronary venous blood. Coronary vascular conductance was calculated as the ratio of normalized CBF to MAP. Coronary arteriovenous [H+] gradient was calculated and presented as coronary venous [H+] minus arterial [H+]. Cardiac work was calculated as the product of stroke volume, heart rate, and systolic aortic blood pressure divided by the left ventricular weight. Body oxygen consumption was normalized to body weight. Percent maximal dilation of isolated arterioles was calculated as: [(Dd − Db)/(Dmax − Db) × 100], where Dd is the diameter after a drug concentration, Db is baseline diameter, and Dmax is maximal passive diameter. The statistical analysis was performed using two-way ANOVA followed by Bonferroni post hoc testing, when appropriate, and using linear regression. Phenotype data between strains was analyzed by unpaired t tests. A p value <0.05 was considered significant. Data are expressed as mean ± SE.
Results
Phenotype of control and FH swine
Phenotypic characteristics of control and FH swine are presented in Table 1. FH swine had higher body and heart weights, but a lower heart weight-to-body weight ratio compared to control swine. As expected, FH swine exhibited significant elevations in total cholesterol, but not triglycerides or glucose, compared to control swine. We have previously reported that the elevation in total cholesterol in this model is due primarily to dramatic increases in low-density lipoprotein cholesterol with a modest elevation of high-density lipoprotein cholesterol [2, 8]. FH swine exhibited diffuse, but not critical (i.e., >70 % luminal narrowing), atherosclerosis burden along the LAD, assessed by IVUS (Fig. 1).

Coronary atherosclerosis in the left anterior descending (LAD) coronary artery of FH swine, assessed by IVUS. Atherosclerosis burden (percent vessel volume reduction) was assessed in proximal (Prox), mid, and distal LAD in FH swine. Representative IVUS pullback of the LAD coronary artery (right panel) with discrimination of the vessel wall (green) and luminal border of the atherosclerotic plaque (red). Values are mean ± SE
Coronary vasodilator function in control and FH swine
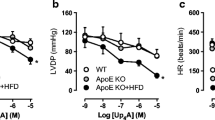
Coronary vasodilator function was assessed via changes in coronary vascular conductance (CVC) in response to the infusion of endothelium-dependent (ATP) and -independent (SNP and bimakalim) vasodilators. Systemic hemodynamic responses to drug infusions were not different between groups (Table 2). Surprisingly, ATP-induced coronary vasodilation was not different between control and FH swine (Fig. 2a). However, coronary vasodilation to SNP, an index of smooth muscle NO sensitivity, was increased in FH swine compared to control (Fig. 2b), suggesting that a reduced bioavailability of NO was compensated for by increased smooth muscle sensitivity to NO. Vasodilation in response to the KATP channel opener bimakalim was not different between these two strains (Fig. 2c).

In vivo coronary vasodilator responses in control and FH swine. Percent increases in coronary vascular conductance (CVC) (i.e., coronary vasodilation) were assessed during infusion of a endothelium-dependent vasodilator ATP, b endothelium-independent nitric oxide donor sodium nitroprusside (SNP), and c endothelium-independent ATP-sensitive potassium channel (KATP) opener bimakalim. Values are mean ± SE, n = 4–7, *p < 0.05 main effect FH versus control
Endothelial dysfunction with increased smooth muscle NO sensitivity in FH was confirmed in isolated arterioles demonstrating reduced vasodilation to the endothelium-dependent dilator BK (Fig. 3a) and increased dilation to SNP (Fig. 3b) in FH. Maximum passive internal diameters were similar for arterioles from control and FH swine (102 ± 4 versus 110 ± 7 µm).

In vitro coronary arteriolar vasodilator responses in control and FH swine. Vasodilation of isolated, pressurized coronary arterioles was determined to bradykinin and SNP. Values are mean ± SE, n = 41 (Con bradykinin), 10 (FH bradykinin), 16 (Con SNP), and 5 (FH SNP), *p < 0.05 main effect FH versus control
Myocardial O2 balance in control and FH swine
The impact of FH on myocardial O2 balance was assessed during graded treadmill exercise in control and FH swine. Treadmill exercise increased heart rate, systolic arterial pressure, and body oxygen consumption per kg body weight at comparable treadmill speeds (2 and 3 mph) in control and FH swine (Table 3). FH swine, in general, would not exercise beyond 3 mph and exhibited increased left atrial pressures at rest and during exercise (Table 3), as previously reported [2, 8].
Exercise-induced increases in myocardial O2 demand (i.e., MVO2 per gram of myocardium) in control swine were slightly exceeded by the increase in CBF g−1 (Tables 3, 4), allowing a small decrease in myocardial O2 extraction (MO2ex), resulting in small increases in SO2cv and PO2cv (Fig. 4). This observation is consistent with the lack of significant α-adrenergic control of the porcine coronary microcirculation [11, 47]. Myocardial arteriovenous H+ concentration gradient remained relatively constant during exercise. Coronary venous (cv) O2 content increased in control swine during exercise likely due to the combined effect of increased CBF g−1, increased hematocrit (i.e., arterial Hb; Tables 3, 4), and the small reduction in MO2ex (Fig. 4).

In vivo coronary resistance vessel tone in control and FH swine. Shown are relations between myocardial oxygen consumption per gram of myocardium (MVO2) and a myocardial oxygen extraction (MO2ex), b coronary venous oxygen saturation (SO2cv) and c tension (PO2cv), d coronary venous oxygen content, and e myocardial arteriovenous H+ concentration gradient (Δ [H+] cv-art). The arteriovenous H+ concentration gradient correlates with mid-LAD plaque burden (percent vessel volume reduction) in FH swine, assessed by IVUS (f). Values are mean ± SE, *p < 0.05 parallel shift versus control, **p < 0.1 parallel shift versus control, † p < 0.05 rotation versus control
Resting MVO2 g−1 was similar between FH and control swine (Table 3) and, in general, FH swine exhibited increased MO2ex, reduced SO2cv, and reduced coronary venous O2 content compared to control swine (Fig. 4). During exercise, FH swine had higher MVO2 g−1 compared to control swine at 3 mph (Table 3) that was matched by a similarly elevated CBF g−1 (Table 3) such that MO2ex, SO2cv, PO2cv, and coronary venous O2 content remained constant during exercise (Fig. 4). Importantly, and in contrast to control swine, FH swine exhibited an increase in the myocardial arteriovenous H+ concentration gradient at 3 mph consistent with a shift toward anaerobic metabolism due to cardiac ischemia (Table 4; Fig. 4). Further analysis revealed that the myocardial arteriovenous H+ concentration gradient at 3 mph in FH swine correlated with the plaque burden of the mid-LAD coronary artery (Fig. 4).
Regional myocardial blood flows in control and FH swine
To assess the impact of FH on the transmural distribution of myocardial blood flows at rest and during exercise, we employed the microsphere technique. The transmural distribution of blood flow at rest and during exercise and the corresponding subendocardial-to-subepicardial blood flow ratios in control and FH swine are shown in Fig. 5a. Resting myocardial blood flows were elevated in FH compared to control swine, but FH swine exhibited a normal subendocardial-to-subepicardial blood flow ratio at rest. During exercise, myocardial blood flow rates across the LV free wall increased to reach similar levels in control and FH swine resulting in similar subendocardial-to-subepicardial blood flow ratios in both strains during exercise (Fig. 5b).

Myocardial blood flow per gram of myocardium in four layers across the left ventricular free wall at rest and during exercise in control and FH swine. a Blood flow to the subepicardium (Epi), outer mid (OM) myocardium, inner mid (IM) myocardium, and subendocardium (Endo) were determined by the infusion of microspheres at rest and during treadmill exercise at 3–4 mph. b Subendocardial-to-subepicardial blood flow ratio (Endo/Epi) of the left ventricular free wall in control and FH swine at rest and during exercise at 3–4 mph. Values are mean ± SE, *p < 0.05 versus Con Rest, † p < 0.05 versus Rest within group
Cardiac function in control and FH swine
FH swine exhibited relatively higher MVO2 than control swine, especially at 3 mph (Table 4), so we further examined the relation between cardiac work and MVO2 (i.e., cardiac efficiency). FH swine exhibited reduced cardiac efficiency compared to control swine that was exacerbated during exercise (Fig. 6a). Our data indicate that FH hearts consume more O2 at a given level of work than control hearts, which is only partly met by an increase in coronary perfusion and O2 delivery (Fig. 4). These perturbations were accompanied by a lower stroke volume (indexed to body weight; Fig. 6b) and elevated left atrial pressure in FH swine (Table 3; Fig. 6c).

Cardiac function in control and FH swine. Shown are relations between myocardial oxygen consumption (indexed to myocardial weight) and a cardiac work (indexed to left ventricular weight) as well as relations between body oxygen consumption (indexed to body weight) and b stroke volume (indexed to body weight) and c left atrial pressure. Values are mean ± SE, n = 4–7, *p < 0.05 parallel shift versus control, † p < 0.05 rotation versus control
Discussion
This study examined whether FH impairs coronary blood flow regulation and myocardial oxygen delivery. The principle findings of this study are that (1) FH reduced coronary endothelial function due to a reduction in NO bioavailability that was compensated by an increase in coronary vascular smooth muscle NO sensitivity; (2) FH demonstrated perturbations in myocardial oxygen balance that were accompanied by a shift toward cardiac anaerobic metabolism, particularly during exercise, indicated by an increased coronary arteriovenous H+ concentration gradient; (3) these abnormalities in oxygen supply were not associated with an alteration in the transmural distribution of myocardial blood flow across the left ventricular wall; however, (4) they were associated with a generalized impairment in cardiac contractile efficiency that worsened during exercise in FH.
An association between impaired coronary microvascular function and major adverse cardiovascular events was recently reported in patients with “intermediate” coronary artery stenosis (40–70 % luminal narrowing) [52]. This study and others [6, 13, 37, 41, 43] have challenged the stenosis-centered paradigm of cardiac ischemia in hypercholesterolemia and FH and highlight a growing appreciation for the role of coronary microvascular dysfunction. Indeed, evidence of impaired coronary microvascular function in hypercholesterolemia prior to the development of critical coronary stenosis has been reported [10, 55]. Our results are consistent with these previous reports by demonstrating impaired coronary endothelial function in vivo and in vitro in FH swine with “intermediate” coronary artery stenosis. Specifically, FH resulted in increased coronary smooth muscle sensitivity to NO (i.e., increased dilation to SNP) in vivo and in vitro. This was coupled with maintained endothelium- and NO-dependent coronary dilation to ATP in vivo and a rightward shifted dilation to bradykinin with maintained maximal dilation (i.e., reduced sensitivity) in vitro in isolated arterioles. These results may suggest an impairment of endothelium-derived hyperpolarizing factor (EDHF)-induced coronary vasodilation in FH, since bradykinin-induced coronary vasodilation in vitro involves EDHF and NO [26], whereas at the doses examined, ATP-induced coronary dilation in vivo is NO-dependent [14]. Both our in vivo and in vitro results imply that NO-dependent vasodilation is maintained in FH swine by a compensatory increase in NO sensitivity, similar to our previous report in the systemic vasculature of FH swine [8].
Our findings extend prior work by demonstrating that FH alters myocardial O2 balance at rest and during exercise. Thus, FH swine demonstrated high O2 consumption for a similar level of cardiac work at rest compared to control swine, which was exacerbated during exercise in FH thereby causing a rotation in the relation between cardiac work and MVO2. This reduced cardiac efficiency in FH may have resulted from impaired cardiac mitochondrial function. Indeed, cardiac mitochondrial uncoupling has been reported in a similar swine model of moderate coronary atherosclerosis [36] and increased cardiac mitochondrial oxidative stress and enhanced mitochondrial permeability transition pore response has been reported in FH swine [33]. A recent report proposed a critical link between cardiac mitochondrial dysfunction and perturbations in coronary microvascular function in metabolic disease [23]. Consistent with those observations, the present study shows that a higher O2 consumption in FH swine was not fully met by a commensurate increase in O2 delivery, necessitating an increase in myocardial O2 extraction thereby leading to reduced coronary venous PO2 and SO2 compared to control swine. The extent of the reduction in coronary venous SO2 is comparable to that reported previously during exercise in swine with chronic myocardial infarction [12] or metabolic syndrome [5], and reflects perturbations in the regulation of coronary resistance vessel tone [11]. These perturbations were accompanied by a shift toward anaerobic cardiac metabolism (i.e., widening of the coronary arteriovenous H+ concentration gradient) in FH, an indicator of suboptimal matching of perfusion to the increased MVO2. These data support a role for impaired myocardial O2 balance as a contributor to impaired cardiac function in FH, especially during exercise.
Our data revealed a strong relationship between mid-LAD plaque burden and widening coronary arteriovenous H+ concentration gradient in FH swine during exercise. Given the diffuse nature of atherosclerosis in this model and the potential exacerbation of stenosis severity during exercise [11], we examined transmural myocardial blood flow distribution via microsphere infusion which revealed no shift in the subendocardial-to-subepicardial distribution of coronary blood flow during exercise. Exacerbated stenosis severity during exercise would be expected to cause subendocardial hypoperfusion due to the exercise-induced increase in average subendocardial tissue pressure [1, 11]. Thus, we do not view the relationship between plaque burden and the coronary arteriovenous H+ concentration gradient as cause-and-effect but rather indicative of similarity in the extent of macrovascular and microvascular disease in this model.
The specific microvascular mechanism(s) in this model are unclear but several observations warrant further investigation. First, microvascular/capillary rarefaction has been reported in skeletal and cardiac muscle in several rodent models of hypercholesterolemia and metabolic syndrome [20, 22, 39, 49]. In skeletal muscle of the obese Zucker rat, microvascular density was inversely proportionate to plasma cholesterol concentration, proportionate to vascular NO bioavailability, and rarefaction was partially prevented by antioxidant treatment to increase NO bioavailability [19, 22]. Thus, FH-associated reductions in NO bioavailability may lead to coronary microvascular rarefaction as a mechanism underlying impaired O2 supply in this condition. Second, hypercholesterolemia has been shown to blunt adenosine-induced vasodilation [25], a known mechanism of coronary arteriolar dilation in hypoperfused myocardium [11] and a clinically relevant agent for the determination of coronary flow reserve [27, 38]. Thus, while our myocardial blood flow data do not demonstrate selective subendocardial hypoperfusion during exercise, FH swine may exhibit diffuse impairment of adenosine responsiveness contributing to diffuse myocardial ischemia at high MVO2. Future studies are warranted to explore the potential contribution of these mechanisms to impaired myocardial O2 balance and exercise-induced ischemia in FH.
A central role for co-morbidity-induced microvascular dysfunction underlying the development of cardiac diastolic dysfunction has recently been proposed. Indeed, although independent of risk factors, a link between impaired coronary O2 delivery and impaired cardiac ventricular function has been reported in chronically instrumented dogs [16] and swine [24]. In the present study, FH swine exhibited a significant impairment of cardiac efficiency and stroke volume as well as increased left atrial pressures. While our data suggest a role for coronary microvascular dysfunction in impaired cardiac function in FH, the progression of this dysfunction remains uncertain. Specifically, whether stroke volume is low due to diastolic dysfunction (i.e., due to restricted filling), systolic dysfunction (i.e., due to impaired contractility with maintained high wall stress/MVO2), or a combination of these requires further examination of cardiac function in this model.
Taken together, our data reveal FH-associated coronary microvascular dysfunction as a primary contributor to impaired myocardial O2 balance and cardiac efficiency prior to the development of critical coronary stenosis. These data provide insight into potential causes of the substantial risk of major adverse cardiovascular events in patients with “intermediate” coronary artery stenosis by highlighting a role for myocardial ischemia and worsening of cardiac function, particularly during increases in cardiac metabolism (i.e., exercise). Thus, we hypothesize that therapeutic targeting of hypercholesterolemia and/or other associated causes of coronary microvascular dysfunction (i.e., inflammation and oxidative stress) could improve cardiovascular outcomes in patients with FH.
References
Ball RM, Bache RJ (1976) Distribution of myocardial blood flow in the exercising dog with restricted coronary artery inflow. Circ Res 38:60–66. doi:10.1161/01.res.38.2.60
Bender SB, de Beer VJ, Tharp DL, van Deel ED, Bowles DK, Duncker DJ, Laughlin MH, Merkus D (2014) Reduced contribution of endothelin to the regulation of systemic and pulmonary vascular tone in severe familial hypercholesterolaemia. J Physiol 592:1757–1769. doi:10.1113/jphysiol.2013.267351
Bender SB, Tune JD, Borbouse L, Long X, Sturek M, Laughlin MH (2009) Altered mechanism of adenosine-induced coronary arteriolar dilation in early-stage metabolic syndrome. Exp Biol Med (Maywood) 234:683–692. doi:10.3181/0812-RM-350
Bender SB, van Houwelingen MJ, Merkus D, Duncker DJ, Laughlin MH (2010) Quantitative analysis of exercise-induced enhancement of early and late systolic retrograde coronary blood flow. J Appl Physiol 108:507–514. doi:10.1152/japplphysiol.01096.2009
Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD (2012) Contribution of voltage-dependent K+ channels to metabolic control of coronary blood flow. J Mol Cell Cardiol 52:912–919. doi:10.1016/j.yjmcc.2011.07.004
Camici PG, d’Amati G, Rimoldi O (2015) Coronary microvascular dysfunction: mechanisms and functional assessment. Nat Rev Cardiol 12:48–62. doi:10.1038/nrcardio.2014.160
Checovich WJ, Fitch WL, Krauss RM, Smith MP, Rapacz J, Smith CL, Attie AD (1988) Defective catabolism and abnormal composition of low-density lipoproteins from mutant pigs with hypercholesterolemia. Biochemistry 27:1934–1941. doi:10.1021/bi00406a020
de Beer VJ, Merkus D, Bender SB, Tharp DL, Bowles DK, Duncker DJ, Laughlin MH (2013) Familial hypercholesterolemia impairs exercise-induced vasodilation due to reduced NO bioavailability. J Appl Physiol 115:1767–1776. doi:10.1152/japplphysiol.00619.2013
de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC (2016) Prevalence of familial hypercholesterolemia in the 1999–2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 133:1067–1072. doi:10.1161/circulationaha.115.018791
Drexler H, Zeiher AM (1991) Endothelial function in human coronary arteries in vivo. Focus on hypercholesterolemia. Hypertension 18:II90–II99. doi:10.1161/01.HYP.18.4_Suppl.II90
Duncker DJ, Bache RJ (2008) Regulation of coronary blood flow during exercise. Physiol Rev 88:1009–1086. doi:10.1152/physrev.00045.2006
Duncker DJ, Beer VJ, Merkus D (2008) Alterations in vasomotor control of coronary resistance vessels in remodelled myocardium of swine with a recent myocardial infarction. Med Biol Eng Comput 46:485–497. doi:10.1007/s11517-008-0315-1
Duncker DJ, Koller A, Merkus D, Canty JM Jr (2015) Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis 57:409–422. doi:10.1016/j.pcad.2014.12.002
Duncker DJ, Stubenitsky R, Tonino PA, Verdouw PD (2000) Nitric oxide contributes to the regulation of vasomotor tone but does not modulate O2-consumption in exercising swine. Cardiovasc Res 47:738–748. doi:10.1016/S0008-6363(00)00143-7
Duncker DJ, Stubenitsky R, Verdouw PD (1998) Role of adenosine in the regulation of coronary blood flow in swine at rest and during treadmill exercise. Am J Physiol 275:H1663–H1672
Farhi ER, Canty JM, Klocke FJ (1989) Effects of graded reductions in coronary perfusion pressure on the diastolic pressure-segment length relation and the rate of isovolumic relaxation in the resting conscious dog. Circulation 80:1458–1468. doi:10.1161/01.cir.80.5.1458
Fleenor BS, Bowles DK (2009) Exercise training decreases the size and alters the composition of the neointima in a porcine model of percutaneous transluminal coronary angioplasty (PTCA). J Appl Physiol 107:937–945. doi:10.1152/japplphysiol.91444.2008
Fleenor BS, Bowles DK (2009) Negligible contribution of coronary adventitial fibroblasts to neointimal formation following balloon angioplasty in swine. Am J Physiol Heart Circ Physiol 296:H1532–H1539. doi:10.1152/ajpheart.00566.2008
Frisbee JC (2005) Reduced nitric oxide bioavailability contributes to skeletal muscle microvessel rarefaction in the metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 289:R307–R316
Frisbee JC, Goodwill AG, Butcher JT, Olfert IM (2011) Divergence between arterial perfusion and fatigue resistance in skeletal muscle in the metabolic syndrome. Exp Physiol 96:369–383. doi:10.1113/expphysiol.2010.055418
Goldberg AC, Gidding SS (2016) Knowing the prevalence of familial hypercholesterolemia matters. Circulation 133:1054–1057. doi:10.1161/circulationaha.116.021673
Goodwill AG, Frisbee SJ, Stapleton PA, James ME, Frisbee JC (2009) Impact of chronic anticholesterol therapy on development of microvascular rarefaction in the metabolic syndrome. Microcirculation 16:667–684. doi:10.3109/10739680903133722
Guarini G, Kiyooka T, Ohanyan V, Pung YF, Marzilli M, Chen YR, Chen CL, Kang PT, Hardwick JP, Kolz CL, Yin L, Wilson GL, Shokolenko I, Dobson JG, Fenton R, Chilian WM (2016) Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res Cardiol 111:1–13. doi:10.1007/s00395-016-0547-4
Guth BD, Wisneski JA, Neese RA, White FC, Heusch G, Mazer CD, Gertz EW (1990) Myocardial lactate release during ischemia in swine. Relation to regional blood flow. Circulation 81:1948–1958. doi:10.1161/01.cir.81.6.1948
Heaps CL, Tharp DL, Bowles DK (2005) Hypercholesterolemia abolishes voltage-dependent K+ channel contribution to adenosine-mediated relaxation in porcine coronary arterioles. Am J Physiol Heart Circ Physiol 288:H568–H576. doi:10.1152/ajpheart.00157.2004
Henderson KK, Turk JR, Rush JW, Laughlin MH (2004) Endothelial function in coronary arterioles from pigs with early-stage coronary disease induced by high-fat, high-cholesterol diet: effect of exercise. J Appl Physiol 97:1159–1168. doi:10.1152/japplphysiol.00261.2004
Heusch G (2010) Adenosine and maximum coronary vasodilation in humans: myth and misconceptions in the assessment of coronary reserve. Basic Res Cardiol 105:1–5. doi:10.1007/s00395-009-0074-7
Kilroy JP, Klibanov AL, Wamhoff BR, Bowles DK, Hossack JA (2014) Localized in vivo model drug delivery with intravascular ultrasound and microbubbles. Ultrasound Med Biol 40:2458–2467. doi:10.1016/j.ultrasmedbio.2014.04.007
Kuo L, Davis MJ, Cannon MS, Chilian WM (1992) Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation. Restoration of endothelium-dependent responses by l-arginine. Circ Res 70:465–476. doi:10.1161/01.RES.70.3.465
Lario FC, Miname MH, Tsutsui JM, Santos RD, Kowatsch I, Sbano JCN, Ramires JAF, Filho RK, Mathias W (2013) Atorvastatin treatment improves myocardial and peripheral blood flow in familial hypercholesterolemia subjects without evidence of coronary atherosclerosis. Echocardiography 30:64–71. doi:10.1111/j.1540-8175.2012.01810.x
Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM (2011) Familial hypercholesterolemia: epidemiology, neolithic origins and modern geographic distribution. Crit Rev Clin Lab Sci 48:1–18. doi:10.3109/10408363.2011.565585
Lowe SW, Checovich WJ, Rapacz J, Attie AD (1988) Defective receptor binding of low density lipoprotein from pigs possessing mutant apolipoprotein B alleles. J Biol Chem 263:15467–15473
McCommis KS, McGee AM, Laughlin MH, Bowles DK, Baines CP (2011) Hypercholesterolemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium: beneficial effects of chronic exercise. Am J Physiol Regul Integr Comp Physiol 301:R1250–R1258. doi:10.1152/ajpregu.00841.2010
Merkus D, Sorop O, Houweling B, Boomsma F, van den Meiracker AH, Duncker DJ (2006) NO and prostanoids blunt endothelin-mediated coronary vasoconstrictor influence in exercising swine. Am J Physiol Heart Circ Physiol 291:H2075–H2081. doi:10.1152/ajpheart.01109.2005
Merkus D, Sorop O, Houweling B, Hoogteijling BA, Duncker DJ (2006) KCa channels contribute to exercise-induced coronary vasodilation in swine. Am J Physiol Heart Circ Physiol 291:H2090–H2097. doi:10.1152/ajpheart.00315.2006
Morrison ES, Scott RF, Lee WM, Frick J, Kroms M, Cheney CP (1977) Oxidative phosphorylation and aspects of calcium metabolism in myocardia of hypercholesterolaemic swine with moderate coronary atherosclerosis. Cardiovasc Res 11:547–553. doi:10.1093/cvr/11.6.547
Murthy VL, Naya M, Foster CR, Gaber M, Hainer J, Klein J, Dorbala S, Blankstein R, Di Carli MF (2012) Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 126:1858–1868. doi:10.1161/CIRCULATIONAHA.112.120402
Naoumova RP, Kindler H, Leccisotti L, Mongillo M, Khan MT, Neuwirth C, Seed M, Holvoet P, Betteridge J, Camici PG (2007) Pioglitazone improves myocardial blood flow and glucose utilization in nondiabetic patients with combined hyperlipidemia—a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol 50:2051–2058. doi:10.1016/j.jacc.2007.07.070
Nascimento AR, Machado M, de Jesus N, Gomes F, Lessa MA, Bonomo IT, Tibiriçá E (2013) Structural and functional microvascular alterations in a rat model of metabolic syndrome induced by a high-fat diet. Obesity 21:2046–2054. doi:10.1002/oby.20358
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AFH, Stroes E, Taskinen M-R, Tybjærg-Hansen A (2013) Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Consensus Statement of the European Atherosclerosis Society. Eur Heart J 34:3478–3490. doi:10.1093/eurheartj/eht273
Paulus WJ, Tschöpe C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271. doi:10.1016/j.jacc.2013.02.092
Prescott MF, McBride CH, Hasler-Rapacz J, Von Linden J, Rapacz J (1991) Development of complex atherosclerotic lesions in pigs with inherited hyper-LDL cholesterolemia bearing mutant alleles for apolipoprotein B. Am J Pathol 139:139–147
Pries AR, Badimon L, Bugiardini R, Camici PG, Dorobantu M, Duncker DJ, Escaned J, Koller A, Piek JJ, de Wit C (2015) Coronary vascular regulation, remodelling, and collateralization: mechanisms and clinical implications on behalf of the working group on coronary pathophysiology and microcirculation. Eur Heart J 36:3134–3146. doi:10.1093/eurheartj/ehv100
Rapacz J, Hasler-Rapacz J, Taylor KM, Checovich WJ, Attie AD (1986) Lipoprotein mutations in pigs are associated with elevated plasma cholesterol and atherosclerosis. Science 234:1573–1577. doi:10.1126/science.3787263
Reinhardt CP, Dalhberg S, Tries MA, Marcel R, Leppo JA (2001) Stable labeled microspheres to measure perfusion: validation of a neutron activation assay technique. Am J Physiol Heart Circ Physiol 280:H108–H116
Rodriguez-Porcel M, Lerman LO, Herrmann J, Sawamura T, Napoli C, Lerman A (2003) Hypercholesterolemia and hypertension have synergistic deleterious effects on coronary endothelial function. Arterioscler Thromb Vasc Biol 23:885–891. doi:10.1161/01.atv.0000069209.26507.bf
Schulz R, Oudiz RJ, Guth BD, Heusch G (1990) Minimal α1- and α2-adrenoceptor-mediated coronary vasoconstriction in the anaesthetized swine. Naunyn Schmiedebergs Arch Pharmacol 342:422–428. doi:10.1007/bf00169459
Shioiri H, Komaru T, Sato K, Takahashi K, Takeda S, Kanatsuka H, Watanabe J, Shirato K (2003) Impact of hypercholesterolemia on acidosis-induced coronary microvascular dilation. Basic Res Cardiol 98:76–83. doi:10.1007/s00395-003-0391-1
Stapleton PA, Goodwill AG, James ME, D’Audiffret AC, Frisbee JC (2010) Differential impact of familial hypercholesterolemia and combined hyperlipidemia on vascular wall and network remodeling in mice. Microcirculation 17:47–58. doi:10.1111/j.1549-8719.2009.00003.x
Tharp DL, Masseau I, Ivey J, Ganjam VK, Bowles DK (2009) Endogenous testosterone attenuates neointima formation after moderate coronary balloon injury in male swine. Cardiovasc Res 82:152–160. doi:10.1093/cvr/cvp038
Tharp DL, Wamhoff BR, Wulff H, Raman G, Cheong A, Bowles DK (2008) Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol 28:1084–1089. doi:10.1161/atvbaha.107.155796
van de Hoef TP, van Lavieren MA, Damman P, Delewi R, Piek MA, Chamuleau SAJ, Voskuil M, Henriques JPS, Koch KT, de Winter RJ, Spaan JAE, Siebes M, Tijssen JGP, Meuwissen M, Piek JJ (2014) Physiological basis and long-term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circ Cardiovasc Interv 7:301–311. doi:10.1161/circinterventions.113.001049
Winnik S, Gaul DS, Siciliani G, Lohmann C, Pasterk L, Calatayud N, Weber J, Eriksson U, Auwerx J, van Tits LJ, Lüscher TF, Matter CM (2016) Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: protective role of a novel C/EBP-β-dependent feedback regulation of SOD2. Basic Res Cardiol 111:1–15. doi:10.1007/s00395-016-0552-7
Yokoyama I, Murakami T, Ohtake T, Momomura S-i, Nishikawa J, Sasaki Y, Omata M (1996) Reduced coronary flow reserve in familial hypercholesterolemia. J Nucl Med 37:1937–1942
Zeiher AM, Drexler H, Wollschläger H, Just H (1991) Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation 83:391–401. doi:10.1161/01.cir.83.2.391
Acknowledgments
We gratefully acknowledge the assistance of Pam Thorne, Dave Harah, Jan Ivey, Sherrie Neff, Dr. Richard McAllister, Elza D. van Deel, and Rob van Bremen. This work was supported by the National Institutes of Health Grants HL-52490 (to MHL) and AR-048523 (to SBB), European Commission FP9-Health-2010 Grant MEDIA-261409 (to DJD and DM), the Netherlands Cardiovascular Research Initiative: an initiative of the Dutch Heart Foundation [CVON2012-08 PHAEDRA to DM; CVON2014-11 RECONNECT to DM and DJD], and the Department of Veterans Affairs Biomedical Laboratory Research and Development CDA-2 IK2 BX002030 (to SBB). This work was supported with resources and the use of facilities at the Harry S Truman Memorial Veterans Hospital in Columbia, MO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
The manuscript does not contain clinical studies or patient data.
Rights and permissions
About this article

Cite this article
Bender, S.B., de Beer, V.J., Tharp, D.L. et al. Severe familial hypercholesterolemia impairs the regulation of coronary blood flow and oxygen supply during exercise. Basic Res Cardiol 111, 61 (2016). https://doi.org/10.1007/s00395-016-0579-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-016-0579-9