
Abstract
Chronic activation of Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway contributes to vascular inflammation and atherosclerosis by inducing expression of genes involved in cell proliferation, differentiation and migration. We aimed to investigate whether enforced expression of negative regulators, the suppressors of cytokine signaling (SOCS1 and SOCS3), inhibits harmful JAK/STAT-mediated responses and affects atherosclerosis in apolipoprotein E knockout mice. Adenovirus-mediated SOCS1 transgene expression impaired the onset and progression of atherosclerosis without impact on lipid profile, whereas SOCS3 was only effective on early atherosclerosis. Mechanistically, SOCS gene delivery, primarily SOCS1, attenuated STAT1 and STAT3 activation and reduced the expression of STAT-dependent genes (chemokine/chemokine receptors, adhesion molecules, pro-inflammatory cytokines and scavenger receptors) in aortic tissue. Furthermore, atherosclerotic plaques exhibit a more stable phenotype characterized by lower lipids, T cells and M1 macrophages and higher M2 macrophages and collagen. Atheroprotection was accompanied by a systemic alteration of T helper- and T regulatory-related genes and a reduced activation state of circulating monocytes. In vascular smooth muscle cells and macrophages, SOCS gene delivery inhibited cytokine-induced STAT activation, pro-inflammatory gene expression, cell migration and proliferation. In conclusion, targeting SOCS proteins, predominantly SOCS1, to suppress pathological mechanisms involved in atheroma plaque progression and destabilization could be an interesting anti-atherosclerotic strategy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis, a chronic multi-factorial disease of the vascular wall, is one of the leading causes of death in Western countries. Inflammation participates in the atherosclerosis process from the initial stages of plaque formation to the thrombotic complications [15]. Besides lipid accumulation in the arterial intima, increased expression of cytokines, adhesion molecules and chemokines participate in endothelial dysfunction, leukocyte infiltration and vascular inflammation, thereby driving atherosclerotic plaque growth and maturation [2, 36]. Therefore, approaches aimed to suppress pro-inflammatory signaling hold the potential to limit cardiovascular events caused by atherogenesis.
Activation of Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway [12, 20] inducing the expression of inflammatory genes regulates key atherosclerotic processes, such as leukocyte recruitment, migration and proliferation of vascular smooth muscle cells (VSMC), apoptosis and foam cell formation [19, 32]. Different JAK/STAT components have been detected in the heart and also in the inflammatory regions of human atherosclerotic plaques [3, 7, 16, 22, 24, 31]. In experimental models, either gene deficiency or pharmacological inhibition of JAK2, STAT1 and STAT3 prevented atherosclerotic lesion formation [1, 5, 7, 16, 28].
The endogenous regulatory family of suppressors of cytokine signaling (SOCS) controls the magnitude and duration of JAK/STAT signaling through several mechanisms, including JAK inhibition, STAT binding and targeting for proteasomal degradation [12, 32]. SOCS proteins regulate development, homeostasis and disease pathogenesis [34]. Indeed, SOCS expression is found increased in inflamed human tissues [37], making them potential markers of ongoing inflammation. Furthermore, loss- and gain-of-function studies evidenced the important role of SOCS proteins in regulating the course of inflammation in different experimental models [6, 21, 27, 35], including cardiovascular diseases [32]. In particular, SOCS1 and SOCS3 isoforms, which predominantly suppress STAT1 and STAT3 activation, have been linked to a variety of pro-atherogenic molecules including cytokines, lipoproteins, angiotensin II, immune complexes, and high glucose in different cell types [7, 10, 21, 22, 24, 31].
Because SOCS-based strategies to impair pathological JAK/STAT activity might be of interest for the treatment of cardiovascular diseases, the present work investigates the anti-inflammatory and atheroprotective properties of SOCS1 and SOCS3 gene delivery in experimental atherosclerosis and in cultured vascular cells.
Methods
Adenoviral vectors
The cloning and production of recombinant adenoviral constructs encoding mouse SOCS1 (Ad-S1) and SOCS3 (Ad-S3) were previously described [21]. Adenovirus were plaque-purified and propagated in packaging human embryonic kidney 293 cells, then purified and titrated by using Adeno-X Virus Kits (BD Biosciences-Clontech, Palo Alto, CA), as detailed [21]. Empty vector (Ad-null) and green fluorescence protein-expressing adenovirus (Ad-GFP) were used as controls.
Atherosclerotic mouse model
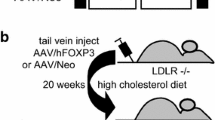
The housing and care of animals and all the procedures carried out in this study were strictly in accordance with the Directive 2010/63/EU of the European Parliament and were approved by the Institutional Animal Care and Use Committee (IIS-Fundacion Jimenez Diaz). Male apolipoprotein E knockout (ApoE KO) mice (C57BL6; Jackson Laboratory, Bar Harbour, ME) aged 8 weeks (early lesion model) or 28 weeks (advanced lesion model) were administered recombinant adenovirus by tail-vein injection (1–2 × 1010 plaque-forming units in 250 μL) and then fed a high-fat Western diet (21 % fat and 0.15 % cholesterol; Harlan Labs, Madison, WI) for 5 weeks. Groups of study: (1) early model: untreated control (n = 15), Ad-null (n = 14), Ad-S1 (n = 16) and Ad-S3 (n = 16); (2) advanced model: untreated control (n = 6), Ad-null (n = 6), Ad-S1 (n = 7) and Ad-S3 (n = 7). At the study endpoint, 16 h-fasted mice were anesthetized (100 mg/kg ketamine and 15 mg/kg xylazine), saline-perfused and killed. Blood was collected by retro-orbital puncture. Serum concentrations of lipids (cholesterol, HDL, LDL and triglycerides) were measured by automated methods. For gene transfer efficiency, mice were studied at 2–14 days after injection (Ad-null, n = 8; Ad-GFP, n = 5; Ad-S1, n = 8; Ad-S3, n = 8).
Flow cytometry
Single-cell suspensions from mouse spleen, femoral bone marrow and EDTA-buffered blood were treated with erythrocyte lysis buffer and then stained with combinations of antibodies to CD45, CD3, CD4, CD8a and CD19 (BD Biosciences) or to CD115 and Ly6C (eBioscience, San Diego, CA). Data were analyzed using BD FACS Canto II Flow Cytometer and BD FACSDiva software (BD Biosciences) [17, 26].
Atherosclerotic lesion analysis
For en face analysis, the aorta was opened longitudinally, stained with Sudan IV and quantified for lipid deposition. To analyze plaque area and composition, aortic root was embedded in optimal cutting temperature medium (Sakura Finetek, Flemingweg, Netherlands) and cryosectioned. Atherosclerotic lesion area (μm2) and neutral lipid content were quantified in 8 μm cross-sections (covering ≈1,000 μm from valve leaflets) after Oil-red-O/hematoxylin staining and averages were calculated from 3–5 sections. Parallel sections were analyzed for collagen content (picrosirius red staining) and immunoperoxidase detection of T lymphocytes (CD4, BD Biosciences), macrophages (Moma2, Serotec, Oxford, UK), and monocyte chemoattractant protein-1 (CCL2, Santa Cruz Biotechnology, Santa Cruz, CA) as described [22]. STAT phosphorylation (P-STAT1, Santa Cruz Biotechnology; P-STAT3, Cell Signaling, Danvers, MA) and macrophage phenotypes (M1, arginase (Arg) II; M2, ArgI; Santa Cruz Biotechnology) were analyzed by immunofluorescence using Alexa Fluor 568 and 488 secondary antibodies (Invitrogen, Carlsbad, CA) followed by nuclear counterstain (4′,6-diamidino-2-phenylindole) [18, 22]. Appropriate antibodies were used as isotype controls. Image acquisition was performed on an inverted epi-fluorescence microscope (Nikon, Melville, NY) with ACT-1 (Nikon) and MetaMorph (Molecular Devices, Sunnyvale, CA) image analysis software. Positive staining was quantified in at least 3 sections per mice (Image-Pro plus, Media Cybernetics, Bethesda, MD) and expressed as percentage or number of positive cells per lesion area [17, 18, 22].
Cell cultures
Mice were euthanized by cervical dislocation under isoflurane anesthesia. Thoracic aortic VSMC isolated by enzymatic digestion (collagenase type II, 290 U/mg; Sigma-Aldrich, St Louis, MO) were cultured in DMEM containing 10 % FBS, 100 U/mL penicillin, 100 µg/mL streptomycin and 2 mM l-glutamine (Life Technologies, Rockville, MD) as reported [18, 22]. Mouse peritoneal macrophages collected by peritoneal lavage were seeded (1 × 107 cells/well) into 6-well culture plates (Corning, NY) and adherent cells were processed [17]. Murine macrophage cell line (RAW 264.7, TIB-71; American Type Culture Collection, Manassas, VA) was maintained in DMEM with 10 % FBS. Cells were incubated with recombinant adenovirus (multiplicity of infection = 40) for 24 h before stimulation (103 U/mL IFNγ plus 102 U/mL IL-6; Peprotech, Rocky Hill, NJ).
mRNA expression analysis
Total RNA from mouse tissues (liver, thoracoabdominal aorta and spleen) and cultured cells was extracted with Tryzol (Life Technologies) [18, 22]. Target gene expression (socs1, socs3, ccl2, ccr2, ccl5, ccr5, icam-1, tnfα, ifnγ, il-4, il-17, foxp3, cd204 and cd36) was analyzed by real-time quantitative PCR (Applied Biosystem, Foster City, CA) and normalized to 18S housekeeping gene.
Protein expression analysis
Cells were lysed in 10 mM Tris pH 7.4, 150 mM NaCl containing 1 % Triton X-100, 0.5 % NP-40, 1 mM EDTA, 1 mM EGTA, 0.2 mM Na3VO4, 10 mM NaF, 0.2 mM PMSF, and protease inhibitor cocktail [17]. Total proteins (20 μg) were electrophoresed and immunoblotted for P-STAT1, P-STAT3, SOCS1, SOCS3 and α-tubulin (Life Technologies and Santa Cruz Biotechnology). Chemokine levels in cell supernatants were measured by ELISA (BD Biosciences and eBioscience).
Luciferase reporter assay
Cells were cotransfected with luciferase reporter vectors (pSTAT1-Luc, pSTAT3-Luc and Renilla control; BD Biosciences) and recombinant adenovirus, as reported [10, 22]. Twenty-four hours after transfection, cells were stimulated with cytokines for additional 16 h. Dual luciferase activity was measured using a luminometer (Berthold Technologies, Bad Wildbad, Germany).
Cell migration and proliferation assays
VSMC migration was measured by the wound-healing assay [18, 26]. Briefly, VSMC monolayers were incubated with adenovirus followed by a wound injury using a plastic pipette tip. Images were collected during the healing period using a phase contrast microscope (Nikon). Wound closure was measured and normalized to time 0 values. Cell proliferation was assessed by methylene blue assay [22, 26].
Statistical analysis
Differences across groups were considered significant at P < 0.05 using one- or two-way ANOVA followed by Bonferroni’s pairwise comparison test (Prism 5, GraphPad Software Inc, La Joya, CA).
Results
SOCS gene delivery inhibits STAT activation in atherosclerotic mice
Transduction efficiency in mouse tissues was evaluated after administration of either SOCS- or GFP-encoding adenovirus to ApoE KO mice. Enhanced SOCS1 and SOCS3 transgene expression was mainly detected in liver, but also in aortic tissue of mice receiving Ad-S1 and Ad-S3, respectively, as compared with empty adenoviral vector (Ad-null; Fig. 1a). Moreover, intense green fluorescence was detected inside the aortic plaques of Ad-GFP transfected mice, predominantly in macrophage-rich areas (Fig. 1b). Time-course experiments also demonstrated sustained SOCS protein expression in mouse aortic tissue even at 14 days after infection (Fig. 1c). Further experiments in ApoE KO mice (early and advanced lesion models) revealed that Ad-S1 and Ad-S3 infection suppressed JAK/STAT activation in atherosclerotic plaques, as assessed by immunofluorescence analysis of STAT1 and STAT3 tyrosine phosphorylation (Fig. 1d, e).

SOCS gene delivery inhibits STAT activation in atherosclerotic mice. a Real-time PCR analysis of SOCS transgene expression in hepatic and aortic tissues of ApoE KO mice at 8 days post-injection of Ad-null, Ad-S1 or Ad-S3. Values normalized to 18S were converted to fold induction vs. Ad-null and expressed as mean ± SEM (n = 3 mice/group). b Representative fluorescence images (n = 2 independent experiments) showing GFP (green, arrows indicate positive cells) and nuclear staining (DAPI, blue) in aortic root sections from mice injected with Ad-null or Ad-GFP. c Representative immunoblots (n = 2 experiments) of SOCS protein expression over time in mouse aorta of Ad-S1 and Ad-S3 injected mice. d Immunofluorescence detection of P-STAT1 (red), P-STAT3 (green) and nuclear staining (blue) in atherosclerotic plaques of Ad-null, Ad-S1 and Ad-S3 mice. Arrows denote immunopositive cells. e Box-and-whisker plot showing quantification of P-STAT1 and P-STAT3 positive cells in early (13 weeks, n = 8–9 mice/group) and advanced (33 weeks, n = 6–7 mice/group) lesions. *P < 0.05, **P < 0.01 and ***P < 0.001 vs. Ad-null
Effects of SOCS transgene expression on atherosclerosis development
The impact of SOCS gene delivery on atherosclerotic plaque progression was explored in ApoE KO mice at both early and advanced stages of lesion development. Quantification of early atherosclerotic lesions in the whole aorta (en face; Fig. 2a) and the aortic sinus (Fig. 2b, e) of young mice (8 weeks old, 5 weeks of high-fat feeding) showed a marked decrease in plaque formation and lipid content in Ad-S1 and Ad-S3 compared with Ad-null group (% decrease: 48 ± 6 and 27 ± 4, respectively; P < 0.05). Remarkably, atherosclerosis assessment in older mice (33 weeks) revealed that only SOCS1 retarded the progression of already developed atherosclerosis, as evidenced by significant reductions of plaque size (Fig. 2c), extension (Fig. 2d) and lipid content (Fig. 2e) in Ad-S1 mice, as compared with Ad-null. In both experimental models, lesion area in a separate group of age-matched uninfected control mice was similar to Ad-null group, indicating that adenoviral infection per se did not affect atherosclerosis. Moreover, no impact on body weight and serum lipid profile was observed by SOCS gene delivery in both early and advanced lesion models (Table 1).

Effect of SOCS gene delivery on atherosclerotic plaque development. a Representative images of aortic lipid accumulation examined by en face Sudan IV staining in early mouse lesions (13 weeks). Quantification of plaque sizes in the different groups (n = 6–8/group). Individual data are expressed as percentage of the total surface area. The mean is depicted as a single horizontal line (P values as indicated). b Representative photomicrographs of aortic root sections (early lesion model) stained with Oil-red-O/hematoxylin and quantitative analysis of individual lesion area (n = 8–9/group). c Representative images and summary of quantification in advanced lesions (33 weeks, n = 6–7/group). d Average of lesion area throughout the aorta in each group (advanced model). Mean ± SEM (*P < 0.05 vs. Ad-null). e Box-and-whisker plot showing quantification of lipid content per lesion area in aortic roots from early and advanced lesions (*P < 0.05 and **P < 0.01 vs. Ad-null)
SOCS gene delivery alters atherosclerotic plaque composition
Histological assessment of early atherosclerotic lesions revealed a significant reduction of T lymphocytes (CD4), macrophages (Moma2), and CCL2 chemokine in the plaques of Ad-S1 and Ad-S3 mice as compared with control and Ad-null groups (Fig. 3a–d). Regression analysis showed correlation of lesion area with CD4, Moma2 and STAT1 staining in the experimental groups (Pearson r values: 0.52, 0.47 and 0.46, respectively; P < 0.05; not shown), thus indicating a more inflammatory component in larger lesions. Additionally, analysis of collagen distribution (picrosirius red staining; Fig. 3a, e) and calculation of collagen-to-lipid ratio in each group (picrosirius red area/Oil-red-O area: Ad-null, 0.39 ± 0.05; Ad-S1, 1.20 ± 0.18, P < 0.001; Ad-S3, 0.86 ± 0.08, P < 0.05; not shown) revealed a more stable plaque phenotype, preferentially in SOCS1 over-expressing mice.

Changes in atherosclerotic plaque composition by SOCS gene delivery. a Representative images of CD4, Moma2 and CCL2 immunoperoxidase staining, picrosirius red staining, and macrophage phenotype immunofluorescence (red, ArgII (M1); green, ArgI (M2); blue, nuclei) in aortic root sections of early lesion model. Box-and-whisker plots, representing the quantitative analysis of T lymphocytes (b), macrophages (c), CCL2 protein (d), collagen content (e) and M1/M2 ratio (f) in the different groups (n = 6–9 mice/group). *P < 0.05, **P < 0.01 and ***P < 0.001 vs. Ad-null
Reduced vascular and systemic inflammation in SOCS over-expressing mice
Given that SOCS gene delivery reduces the intra-plaque macrophage accumulation, we examined the distribution of macrophage polarization markers, namely ArgII (pro-inflammatory classical M1) and ArgI (anti-inflammatory alternative M2). Immunofluorescence analysis revealed that both macrophage phenotypes are present in atherosclerotic lesions, being ArgII (M1) most abundantly expressed in controls groups and ArgI (M2) the predominant macrophage marker in Ad-S1 and Ad-S3 lesions (Fig. 3a, f).
We next investigated whether SOCS-mediated atheroprotection associates with decreased expression of JAK/STAT-dependent genes. Quantitative real-time PCR analysis on aortic tissues from Ad-S1 and Ad-S3 mice revealed a significant reduction in the gene expression of chemokine/chemokine receptor pairs (CCL2/CCR2, CCL5/CCR5), adhesion molecule (ICAM-1), pro-inflammatory cytokines (TNFα, IFNγ) and scavenger receptors (CD204, CD36) as compared with Ad-null and untreated controls (Fig. 4a).

Impact of SOCS transgene expression on plaque and systemic inflammation. a Real-time quantitative PCR analysis of inflammatory factors and scavenger receptors in aortic mouse tissue at 5 weeks post-transfection. Values normalized to 18S are expressed in arbitrary units. b Expression of Th1, Th2, Th17 and Treg pathway genes in splenic samples from mice of the different groups. c Flow cytometry analysis of relative CD115+ monocyte population (Ly6Chigh and Ly6Clow) in spleen, bone marrow and total blood. Data are shown as a box-and-whisker plot with median (n = 5–8/group). *P < 0.05 vs. Ad-null
The systemic expression of representative genes for T helper (Th) and T regulatory (Treg) responses was assessed in spleen samples. As shown in Fig. 4b, Ad-S1 and Ad-S3 mice exhibited a reduced splenic expression of Th1 (IFNγ) and Th17 (IL-17), but not Th2 (IL-4) cytokines. Additionally, SOCS1 resulted in enhanced expression of Treg-associated transcription factor Foxp3 (Fig. 4b).
The impact of SOCS gene delivery on leukocyte formation was examined by flow cytometry analysis of spleen, bone marrow and whole blood. No significant differences in the number of B cells (CD19), T cells (CD3 and CD4/CD8 subgroups) and monocytes (CD115) were observed among the four groups of mice (Supplemental Fig. S1). Interestingly, analysis of monocyte subset distribution revealed decreased CD115+Ly6Chigh and increased CD115+Ly6Clow monocytes in Ad-S1 and Ad-S3 mice when compared with control groups (Fig. 4c). However, this trend only reached statistical significance in peripheral blood, thus indicating that SOCS gene delivery reduces the activation state of circulating monocytes.
SOCS proteins suppress STAT-mediated inflammatory responses in vascular cells
The in vitro effects of adenovirus-mediated SOCS induction were studied on VSMC and macrophages, key cellular constituents of the atherosclerotic lesion that participate actively in plaque development. In primary mouse VSMC, Ad-S1 and Ad-S3 incubation prevented STAT1/STAT3 tyrosine phosphorylation (Fig. 5a) and STAT-regulated gene expression (Fig. 5b) induced by cytokines (IFNγ plus IL6). Similarly, SOCS1 and SOCS3 transgene expression suppressed the STAT1/STAT3 luciferase transcriptional activity in cytokine-stimulated RAW 264.7 macrophages (Fig. 5c), and also inhibited the mRNA expression of scavenger receptors (CD204 and CD36) in peritoneal macrophages (Fig. 5d). Remarkably, only SOCS1 was able to prime macrophages to the anti-inflammatory phenotype, as evidenced by high ArgI (M2 marker) and low ArgII (M1 marker) expression levels (Fig. 5d) and reduced secretion of CCL2 and CCL5 chemokines (Fig. 5e). In addition, SOCS1 transgene expression significantly inhibited VSMC migration (wound-healing assay; Fig. 5f) and suppressed the mitogenic effects of cytokines on VSMC and macrophages (Fig. 5g).

Effects of adenovirus-mediated SOCS expression in cultured cells. Murine VSMC and macrophages were treated with recombinant adenovirus before cytokine stimulation. a Representative immunoblots of P-STAT1/P-STAT3, SOCS1/SOCS3 in VSMC and summary of densitometric analysis (n-fold vs. control conditions). b Real-time PCR analysis of inflammatory genes in VSMC after 6 h of stimulation. c Luciferase reporter assay of STAT1 and STAT3 in RAW 264.7 macrophages. d Real-time PCR analysis of scavenger receptors and M1/M2 markers in peritoneal macrophages. e Secreted CCL2 and CCL5 levels in RAW 264.7 cells at 6 h of stimulation were determined by ELISA. f VSMC migration assessed by wound-healing assays at different time points. g Methylene blue proliferation assay in VSMC and RAW 264.7 macrophages at 24 h of stimulation. Each condition was performed in duplicate or triplicate. Values are mean ± SEM of 4–6 independent experiments. *P < 0.05 vs. basal; #P < 0.05 vs. Ad-null
Discussion
The present study describes that SOCS gene delivery delays the onset and/or progression of atherosclerosis in a mouse model relevant to human atherosclerosis and demonstrates the pivotal role of SOCS family in maintaining a balanced pro- and anti-inflammatory response. Mechanistically, SOCS1 and SOCS3 induction inhibits vascular STAT1/STAT3 activation, reduces the expression of STAT-dependent genes (chemokine/chemokine receptor pairs, adhesion molecules, pro-inflammatory cytokines and scavenger receptors) and therefore affects leukocyte influx, monocyte/macrophage phenotypic balance, VSMC activation, lipid deposition and plaque stability.
Evidence indicates that impairment of SOCS regulatory mechanism is involved in the pathogenesis of immune and inflammatory disorders [12, 34], including cardiovascular diseases [32]. Indeed, SOCS proteins increase locally in acute and chronic inflammatory conditions [37] and SOCS expression in peripheral blood mononuclear cells is proposed as cardiovascular marker in chronic kidney disease patients [25]. Our early studies demonstrated that SOCS1 and SOCS3 proteins are present in atherosclerotic plaques from carotid endarterectomy patients, mainly located in VSMC and macrophages at sites of inflammation [22], and also in kidneys from diabetic patients [21]. Differential SOCS expression in developing lesions of atherosclerotic mice is also reported [14, 22, 33]. Further experimental models demonstrate that inducers/mimics of SOCS proteins delay arthritis development [35], improve survival in lethal endotoxemia [6], impair innate immune responses [27] and also prevent graft arteriosclerosis [24], autoimmune heart disease [30] and the progression of diabetes complications [21, 26]. Conversely, SOCS knockdown, leading to sustained STAT activation, exacerbates inflammatory responses in atherosclerosis-prone models [8, 22]. In line with these studies, our work is the first description of the protective (including anti-inflammatory) effects of adenovirus-mediated SOCS expression in the atherosclerosis context.
Our data indicate that gene delivery of either SOCS1 or SOCS3 impairs STAT1/STAT3 activation in developing atherosclerotic plaques and, strikingly, limits atheroma plaque formation in mice. The underlying mechanisms are independent of changes in serum lipids. Instead, SOCS induction alters the size and composition of atherosclerotic lesions and inhibits plaque inflammation, as revealed by attenuated expression of pro-inflammatory genes involved in recruitment, migration and activation of vascular cells. Remarkably, SOCS gene delivery impairs the accumulation of leukocytes (T cells and macrophages) within the lesions, but also affects the inflammatory state of lesional macrophages. Particularly SOCS1, and to a lesser extent SOCS3, is involved in confining classically activated M1 macrophage formation and fostering alternatively activated M2 macrophage generation. Furthermore, aortic tissue of SOCS over-expressing mice exhibited a reduced expression of CD204 and CD36, two relevant scavenger receptors involved in modified LDL internalization and macrophage foam cell formation [13]. Previous studies have established that M2 macrophages in atherosclerotic plaques display lower lipid-handling capacities and reduced foam cell transformation capacity [4]. Moreover, loss of SOCS1 in the LDL receptor deficient mouse model resulted in enhanced atherosclerotic lesions with accumulation of M1 macrophage foam cells and CD36 expression [8], which is totally compatible with our findings in SOCS transfected mice. Concomitantly, developing atherosclerotic lesions of SOCS over-expressing mice display less lipid and higher collagen content compared with control groups, thereby confirming a less inflamed, more stable plaque phenotype. Considering that most acute complications of atherosclerosis are caused by the rupture of an unstable (leukocyte- and lipid-rich, collagen-poor) plaque [9], efficient SOCS-based strategies to modulate JAK/STAT-dependent responses could be of benefit in slowing lesion progression.
It is also noteworthy to mention that unlike SOCS1, which has a major anti-inflammatory role in both early and advanced atherosclerotic lesions, SOCS3 gene delivery was only effective in the initial phases of atherogenesis. This paradoxical effect of SOCS3 on atherosclerosis regulation could be mostly due to a dual function of STAT3, the transcription factor mainly modulated by SOCS3 [12, 28, 32]. In fact, STAT3 is reported to induce both pro- and anti-inflammatory cytokines, and also to suppress nuclear factor-κB activation and inflammatory gene expression [32]. In the heart, STAT3 signaling contributes to cardioprotection after ischemia/reperfusion injury by promoting cardiomyocyte survival [3]. Furthermore, our previous research demonstrated that antisense oligodeoxynucleotides targeting SOCS3 exacerbate the early atherosclerotic process in ApoE KO mice by increasing lesion size, leukocyte content, and chemokine production [22]. By contrast, T lymphocyte-specific deficiency in SOCS3 reduces atherosclerosis and vascular inflammation in mice [31]. Therefore, it is conceivable that SOCS3 may play multiple roles at later stages of atherosclerosis, the reflected net effect involving a more complex mechanism, all of which require further validation studies.
In the current study we demonstrate that intravenous adenoviral gene delivery is an effective in vivo method to elucidate the role of SOCS in atherogenesis. Injection of SOCS1- and SOCS3-encoding adenovirus resulted in local transgene expression in vessel cells of apoE KO mice, thus suggesting that atheroprotection is mainly derived from a direct action of SOCS on vascular cell activation within the plaques. In vitro data further support the essential role of SOCS proteins in regulating excessive pro-atherogenic activation of VSMC and macrophages, two major cellular constituents of plaques. In both cell types, adenovirus-mediated SOCS1 expression suppresses STAT1, and to a lesser extent STAT3 activation, while SOCS3 predominantly attenuated STAT3 induced by IFNγ and IL-6, two pro-atherogenic cytokines involved in plaque formation and destabilization [2]. Noteworthy, SOCS1 abolished the expression of STAT-regulated genes (e.g. cytokines, chemokines/chemokine receptors, adhesion molecules, and scavenger receptors) which, together with the raise of M2 macrophage marker, resulted in impaired migratory, proliferative and foam formation capacities of vascular cells, all of which have been implicated as underlying mechanisms of atherosclerosis acceleration in vivo.
Besides this local effect on atherosclerotic plaques, we could not exclude the possibility that adenovirus-mediated SOCS transgene expression led to an indirect effect on systemic inflammation. Indeed, we observed that SOCS1, and in a lesser extent SOCS3, reduced the relative number of circulating CD115+Ly6Chigh monocytes, the classical inflammatory subset predominant in different experimental mouse models of atherosclerosis that is preferentially adhered to activated endothelium, accumulated in lesions, and locally differentiated into macrophages [11, 29]. Additionally, splenic changes in the expression of Th- and Treg-associated genes (IFNγ, IL-17 and Foxp3) suggests that SOCS induction affects the outcome of the adaptive immune response at this stage of the disease. Previous studies have established that pro-inflammatory Th1- and Th17-related cytokines promote the development and progression of the disease, whereas anti-inflammatory Th2 and Treg genes exert atheroprotective activities [2, 36, 38]. Evidence also indicates that SOCS1 controls the generation of Th1 and Th2 cells, while SOCS3 modulates Th1 and Th17 polarization [23]. Accordingly, our observations suggest that systemic mechanisms (reduced activation state of circulating monocytes and altered Th1, Th17 and Treg gene expression) also contribute to the in vivo atheroprotective effect of SOCS gene delivery in atherosclerotic mice.
Collectively, our study provide first evidence that systemic delivery of SOCS genes, and predominantly SOCS1, diminishes the atherosclerotic burden and increases plaque stability at two different stages of atherosclerosis in mice. Differential targeting of SOCS family members to impair pathological, pro-inflammatory JAK/STAT activity might provide a therapeutic approach to prevent or retard the course of atherosclerosis.
References
Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM (2007) Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation 115:2939–2947. doi:10.1161/CIRCULATIONAHA.107.696922
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A (2011) Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol 31:969–979. doi:10.1161/ATVBAHA110.207415
Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R (2008) The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 120:172–185. doi:10.1016/j.pharmthera.2008.08.002
Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, Haulon S, Zawadzki C, Jude B, Staels B (2011) Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res 108:985–995. doi:10.1161/CIRCRESAHA.110.233775
Daniel JM, Dutzmann J, Bielenberg W, Widmer-Teske R, Gunduz D, Hamm CW, Sedding DG (2012) Inhibition of STAT3 signaling prevents vascular smooth muscle cell proliferation and neointima formation. Basic Res Cardiol 107:261. doi:10.1007/s00395-012-0261-9
Fang M, Dai H, Yu G, Gong F (2005) Gene delivery of SOCS3 protects mice from lethal endotoxic shock. Cell Mol Immunol 2:373–377
Gharavi NM, Alva JA, Mouillesseaux KP, Lai C, Yeh M, Yeung W, Johnson J, Szeto WL, Hong L, Fishbein M, Wei L, Pfeffer LM, Berliner JA (2007) Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem 282:31460–31468. doi:10.1074/jbc.M704267200
Grothusen C, Schuett H, Hillmer A, Lumpe S, Grote K, Ballmaier M, Bleich A, Glage S, Tietge UJ, Luchtefeld M, Schieffer B (2012) Role of suppressor of cytokine signaling-1 in murine atherosclerosis. PLoS One 7:e51608. doi:10.1371/journal.pone.0051608
Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Oie E, Aukrust P (2008) Atherosclerotic plaque stability—what determines the fate of a plaque? Prog Cardiovasc Dis 51:183–194. doi:10.1016/j.pcad.2008.09.001
Hernandez-Vargas P, Lopez-Franco O, Sanjuan G, Ruperez M, Ortiz-Munoz G, Suzuki Y, Aguado-Roncero P, Perez-Tejerizo G, Blanco J, Egido J, Ruiz-Ortega M, Gomez-Guerrero C (2005) Suppressors of cytokine signaling regulate angiotensin II-activated Janus kinase-signal transducers and activators of transcription pathway in renal cells. J Am Soc Nephrol 16:1673–1683. doi:10.1681/ASN.2004050374
Keul P, Lucke S, von Wnuck LK, Bode C, Graler M, Heusch G, Levkau B (2011) Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res 108:314–323. doi:10.1161/CIRCRESAHA.110.235028
Kiu H, Nicholson SE (2012) Biology and significance of the JAK/STAT signalling pathways. Growth Factors 30:88–106. doi:10.3109/08977194.2012.660936
Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, Morton RE, Febbraio M (2008) Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res 78:185–196. doi:10.1093/cvr/cvm093
Liang X, He M, Chen T, Liu Y, Tian YL, Wu YL, Zhao Y, Shen Y, Yuan ZY (2013) Multiple roles of SOCS proteins: differential expression of SOCS1 and SOCS3 in atherosclerosis. Int J Mol Med 31:1066–1074. doi:10.3892/ijmm.2013.1323
Libby P, Ridker PM, Hansson GK (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473:317–325. doi:10.1038/nature10146
Lim WS, Timmins JM, Seimon TA, Sadler A, Kolodgie FD, Virmani R, Tabas I (2008) Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation 117:940–951. doi:10.1161/CIRCULATIONAHA.107.711275
Mallavia B, Oguiza A, Lopez-Franco O, Recio C, Ortiz-Munoz G, Lazaro I, Lopez-Parra V, Egido J, Gomez-Guerrero C (2013) Gene deficiency in activating fcgamma receptors influences the macrophage phenotypic balance and reduces atherosclerosis in mice. PLoS One 8:e66754. doi:10.1371/journal.pone.0066754
Mallavia B, Recio C, Oguiza A, Ortiz-Munoz G, Lazaro I, Lopez-Parra V, Lopez-Franco O, Schindler S, Depping R, Egido J, Gomez-Guerrero C (2013) Peptide inhibitor of NF-kappaB translocation ameliorates experimental atherosclerosis. Am J Pathol 182:1910–1921. doi:10.1016/j.ajpath.2013.01.022
Marrero MB (2005) Introduction to JAK/STAT signaling and the vasculature. Vascul Pharmacol 43:307–309
Miklossy G, Hilliard TS, Turkson J (2013) Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov 12:611–629. doi:10.1038/nrd4088
Ortiz-Munoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C (2010) Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol 21:763–772. doi:10.1681/ASN.2009060625
Ortiz-Munoz G, Martin-Ventura JL, Hernandez-Vargas P, Mallavia B, Lopez-Parra V, Lopez-Franco O, Munoz-Garcia B, Fernandez-Vizarra P, Ortega L, Egido J, Gomez-Guerrero C (2009) Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler Thromb Vasc Biol 29:525–531. doi:10.1161/ATVBAHA.108.173781
Palmer DC, Restifo NP (2009) Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol 30:592–602. doi:10.1016/j.it.2009.09.009
Qin L, Huang Q, Zhang H, Liu R, Tellides G, Min W, Yu L (2014) SOCS1 prevents graft arteriosclerosis by preserving endothelial cell function. J Am Coll Cardiol 63:21–29. doi:10.1016/j.jacc.2013.08.694
Rastmanesh MM, Bluyssen HA, Joles JA, Boer P, Willekes N, Braam B (2008) Increased expression of SOCS3 in monocytes and SOCS1 in lymphocytes correlates with progressive loss of renal function and cardiovascular risk factors in chronic kidney disease. Eur J Pharmacol 593:99–104. doi:10.1016/j.ejphar.2008.07.013
Recio C, Oguiza A, Lazaro I, Mallavia B, Egido J, Gomez-Guerrero C (2014) Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler Thromb Vasc Biol 34:1953–1960. doi:10.1161/ATVBAHA.114.304144
Sakurai H, Tashiro K, Kawabata K, Yamaguchi T, Sakurai F, Nakagawa S, Mizuguchi H (2008) Adenoviral expression of suppressor of cytokine signaling-1 reduces adenovirus vector-induced innate immune responses. J Immunol 180:4931–4938. doi:10.4049/jimmunol.180.7.4931
Sikorski K, Czerwoniec A, Bujnicki JM, Wesoly J, Bluyssen HA (2011) STAT1 as a novel therapeutical target in pro-atherogenic signal integration of IFNgamma, TLR4 and IL-6 in vascular disease. Cytokine Growth Factor Rev 22:211–219. doi:10.1016/j.cytogfr.2011.06.003
Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117:195–205. doi:10.1172/JCI29950
Tajiri K, Imanaka-Yoshida K, Matsubara A, Tsujimura Y, Hiroe M, Naka T, Shimojo N, Sakai S, Aonuma K, Yasutomi Y (2012) Suppressor of cytokine signaling 1 DNA administration inhibits inflammatory and pathogenic responses in autoimmune myocarditis. J Immunol 189:2043–2053. doi:10.4049/jimmunol.1103610
Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van SJ, Yoshimura A, Tedgui A, Mallat Z (2009) Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med 206:2067–2077. doi:10.1084/jem.20090545
Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A (2011) Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol 31:980–985. doi:10.1161/ATVBAHA.110.207464
Tang J, Kozaki K, Farr AG, Martin PJ, Lindahl P, Betsholtz C, Raines EW (2005) The absence of platelet-derived growth factor-B in circulating cells promotes immune and inflammatory responses in atherosclerosis-prone ApoE-/- mice. Am J Pathol 167:901–912. doi:10.1016/S0002-9440(10)62061-5
Trengove MC, Ward AC (2013) SOCS proteins in development and disease. Am J Clin Exp Immunol 2:1–29
Veenbergen S, Bennink MB, de Hooge AS, Arntz OJ, Smeets RL, van den Berg WB, van de Loo FA (2008) Splenic suppressor of cytokine signaling 3 transgene expression affects T cell responses and prevents development of collagen-induced arthritis. Arthritis Rheum 58:3742–3752. doi:10.1002/art.24072
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17:1410–1422. doi:10.1038/nm.2538
White GE, Cotterill A, Addley MR, Soilleux EJ, Greaves DR (2011) Suppressor of cytokine signalling protein SOCS3 expression is increased at sites of acute and chronic inflammation. J Mol Histol 42:137–151. doi:10.1007/s10735-011-9317-7
Xie JJ, Wang J, Tang TT, Chen J, Gao XL, Yuan J, Zhou ZH, Liao MY, Yao R, Yu X, Wang D, Cheng Y, Liao YH, Cheng X (2010) The Th17/Treg functional imbalance during atherogenesis in ApoE(-/-) mice. Cytokine 49:185–193. doi:10.1016/j.cyto.2009.09.007
Acknowledgments
The authors thank Drs. C.E. Fernandez-Garcia, J.L. Martin-Ventura and L.M. Blanco-Colio (IIS-Fundacion Jimenez Diaz) for their assistance with adenovirus preparation, microscopy and flow cytometry. This work was supported by Grants from Spanish Ministry of Economy and Competitiveness (SAF2012-38830), Ministry of Health (FIS PI10/00072), Iñigo Alvarez de Toledo Renal Foundation and the Spanish Societies of Nephrology and Arteriosclerosis.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Recio and A. Oguiza contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Recio, C., Oguiza, A., Mallavia, B. et al. Gene delivery of suppressors of cytokine signaling (SOCS) inhibits inflammation and atherosclerosis development in mice. Basic Res Cardiol 110, 8 (2015). https://doi.org/10.1007/s00395-014-0458-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-014-0458-1