
Abstract
5-Diphosphoinositol pentakisphosphate (IP7), formed by a family of inositol hexakisphosphate kinases (IP6Ks), has been demonstrated to be a physiologic inhibitor of Akt. IP6K inhibition may increase Akt activation in mesenchymal stem cells (MSCs), resulting in enhanced cardiac protective effect after transplantation. The aim of this study was to investigate the role of IP6Ks for improving MSCs’ functional survival and cardiac protective effect after transplantation into infarcted mice hearts. Bone marrow-derived mesenchymal stem cells, isolated from dual-reporter firefly luciferase and enhanced green fluorescent protein positive (Fluc+–eGFP+) transgenic mice, were preconditioned with IP6Ks inhibitor TNP (0.5, 1, 5, and 10 μmol/L) for 2 h followed by 6 h of hypoxia and serum deprivation (H/SD) injury. TNP concentration dependently significantly decreased IP7 production with increased Akt phosphorylation. Moreover, TNP at 10 μmol/L significantly improved the viability and enhanced the paracrine effect of MSCs after H/SD. Furthermore, MSCs were transplanted into infarcted hearts with or without selective IP6Ks inhibition. Longitudinal in vivo bioluminescence imaging and immunofluorescent staining revealed that TNP pretreatment enhanced the survival of engrafted MSCs, which promoted the anti-apoptotic and pro-angiogenic efficacy of MSCs in vivo. Furthermore, MSC therapy with IP6Ks inhibition significantly decreased fibrosis and preserved heart function. This study demonstrates that inhibition of IP6Ks promotes MSCs engraftment and paracrine effect in infarcted hearts at least in part by down-regulating IP7 production and enhancing Akt activation, which might contribute to the preservation of myocardial function after MI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite a wide range of therapeutic approaches, myocardial infarction (MI) continues to be a major cause of significant morbidity and mortality worldwide [3, 8].Stem cell therapy has emerged as a novel potential additional treatment of ischemic heart disease. Mesenchymal stem cells (MSCs) have been considered an optimal candidate because of their plasticity, ease of isolation and low immunogenicity. However, the strategy of stem cell transplantation is limited by the poor viability and function of donor cells [22, 33]. Therefore, optimizing approaches to augment engrafted cell survival and function is crucial to improving cell therapy for MI.
Genetically modified MSCs over-expressing the survival gene Akt (Akt-MSCs) were markedly resistant to hypoxic injury in vitro and in vivo [15]. The improved survival of Akt-MSCs was associated with an enhanced cardioprotective effect. However, gene transfer approach is not suitable for the clinic because of some potential drawbacks, including insertional mutagenesis, high manufacturing costs, and possible tumorigenicity due to over-expression of Akt [16]. Instead, a pharmaceutical approach involving temporary activation of Akt may be an attractive optimizing strategy to improve the viability of engrafted MSCs.
Inositol phosphates (IPs) are a diverse group of signaling molecules that are widely distributed in mammals [2]. The 5-diphosphoinositol pentakisphosphate (IP7), formed by a family of inositol hexakisphosphate kinases (IP6Ks) including IP6K1, IP6K2, and IP6K3, serves multiple biological functions including inhibiting apoptosis and increasing insulin secretion [23, 24]. Thus far, IP7 has been demonstrated to be a physiologic inhibitor of Akt signaling [6]. Furthermore, a purine analog [N6-(p-nitrobenzyl) purine, TNP] is identified as a reversible inhibitor of IP6Ks, which can concentrate dependently and selectively inhibit IP6K activity [20]. Therefore, TNP provides a means to modulate cellular IP7 synthesis, potentially offering an innovative tool to regulate Akt activity. Accordingly, we hypothesized that IP6K inhibition by TNP could decrease IP7 production and increase Akt activation in MSCs, resulting in enhanced viability and cardiac protective effect after transplantation.
Methods
Detailed methodology is available in the Online Data Supplement.
Animals
Fluc+–eGFP+ transgenic mice [Tg(fluc–egfp)] were bred on a C57BL/6a background to stably express both firefly luciferase Fluc and enhanced green fluorescence protein (eGFP) in all tissues and organs. The experiments were approved by the Fourth Military Medical University Committee on Animal Care.
Hypoxia/serum deprivation injury
MSCs were stimulated with hypoxia/serum deprivation (H/SD) injury as described previously [35]. Briefly, after being replaced in Hanks buffer, MSCs were exposed to hypoxia (94 % N2, 5 %CO2, and 1 % O2) in an anaerobic system (Thermo Forma) at 37 °C for 6 h. In the control group, MSCs were maintained at normoxia (95 % air, 5 % CO2) for equivalent periods.
Radiolabeling and analysis of inositol polyphosphates
To test the IP6Ks activity and cellular concentration of IP7 in MSCs, we monitored inositol polyphosphate levels by high-performance liquid chromatography (HPLC) after labeling with [3H]inositol [13]. In brief, MSCs were seeded onto 6-well culture plates at 1.0 × 104 cells/cm2. After washing with inositol-free medium, cells were cultured with 100 μCi/ml [3H]inositol in DMEM supplemented with 10 % FBS for 72 h. The cells were then scraped after being lysed with 0.5 mol/L trichloroacetic acid. The supernatant was collected and prepared for HPLC analysis. Left ventricles of hearts were dissected and lysed at 4 °C in buffer containing Tris-buffered saline, 0.1 % Triton X-100 (1 mM, Sigma), 4 % glycerol, EDTA (1 mM, Sigma) and protease inhibitor PMSF (1 mM, Roche Molecular Biochemicals). The insoluble material was removed by centrifugation at 6,000×g, and the supernatants were diluted in SDS sample buffer. Finally, the inositol phosphates were eluted by Strong anion exchange high-performance liquid chromatography (SAX-HPLC) with an optimal gradient.
In vitro assessment of TNP's effects on MSCs after H/SD injury
MSCs were stimulated with H/SD injury for 6 h after incubation with different concentrations of TNP (0.5, 1, 5, and 10 μmol/L). The cell viability of MSCs was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and bioluminescence imaging (BLI) using the IVIS Kinetic system as described [5, 35]. MSC apoptosis was determined by flow cytometer assay using an Annexin V-FITC/PI kit (Merck) according to the manufacturer’s instructions. Caspase-3 activity was measured using a caspase-3 assay kit (Abcam) according to the manufacturer’s instructions. The expressions of proteins were evaluated by Western blotting analysis following the standard protocol. The concentrations of VEGF, FGF2, IGF-1, and HGF secreted by MSCs were determined by enzyme-linked immunosorbent assay (ELISA, Life Technologies) [35].
Myocardial infarction mice model and MSC transplantation
Myocardial infarction (MI) was accomplished by ligation of the left anterior descending (LAD) artery [9]. In brief, mice were intubated and ventilated with 50 % oxygen in room air. A left thoracotomy was performed and the pericardium was opened after anesthesia (2 % isoflurane and oxygen). LAD was permanently ligated with a 6.0 suture. The ligation was deemed successful when the anterior wall of the LV turned pale and characteristic ECG changes were recorded. MSCs, cultured in inositol-free medium, were administrated with 10 μmol/L TNP in DMSO for 24 h at 37 °C. For control treatment, 3 μl of DMSO was added to MSCs. The DMSO concentration was 0.1 % of the total volume of medium in all cases. After TNP pre-treatment, the culture medium was aspirated off, and cells were washed with inositol-free medium for three times before collection. TNP pre-treated MSCs (1 × 106) or MSCs (1 × 106) were injected directly into the peri-infarcted areas (multiple injections within the presumed infarct and the border zone) immediately after MI.
In vivo evaluation of MSC engraftment
Bioluminescence imaging (BLI) was performed to track engrafted MSCs using an IVIS® Kinetic system (Caliper, Hopkinton, MA, USA) as described previously [5]. The mice were anesthetized with 2 % isoflurane. After intraperitoneal injection with d-luciferin (375 mg/kg body weight), recipient mice were anesthetized and imaged for 1 min on days 2, 3, 5, 7, 10, 14, 21 until sacrificed. Peak signals (photons/s/cm2/sr) from a fixed region of interest (ROI) were analyzed using Living Image® 4.0 software (Caliper, MA, USA). The survival of MSCs was also confirmed by GFP immunofluorescent stain.
Histological analysis of apoptosis and angiogenesis
Apoptosis in the heart at 48 h after MSCs transplantation was determined by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay as previously described [27]. Fast green/Sirius red stain was performed to detect fibrosis in cardiac muscle in 4 weeks post-procedure. Fibrosis was evaluated by measuring the collagen area as a proportion of the total left ventricular area using the Imaging Pro Plus software. The capillary density was determined by CD31 immunohistochemistry. Vessels in the peri-infarct zone were counted in randomly chosen high-power fields (HPFs, magnification ×400). The results are expressed as vessels per HPF. Immunofluorescence staining of CD31 was performed to visualize CD31-positive vessels. Cell nuclei were stained with DAPI. Sections were imaged using confocal microscope (FluoView-FV1000, Olympus, Japan).
Evaluation of heart function
Echocardiography studies were performed under anesthesia with 2 % isoflurane at 2 days, 7 days, and weekly until sacrificed at 4 weeks post-operation using a 30-MHz transducer on a Vevo® 2100 ultrasound system (VisualSonics, CA). The left ventricular end-systolic volume (LVESV) and left ventricular end-diastolic volume (LVEDV) were measured to calculate left ventricular ejection fraction (LVEF). The left ventricular end-systolic diameter (LVESD) and left ventricular end-diastolic diameter (LVEDD) were measured to calculate left ventricular fractional shortening (FS). For evaluating the LV fractional shortening (FS), endocardial borders were outlined at end-diastole and end-systole, using a left ventricular short-axis view, and FS (%) was calculated by (LV end-diastolic diameter−LV end-systolic diameter)/LV end-diastolic diameter × 100 %. We also evaluated the fractional area shortening and left ventricular anterior wall thickness systolic (LVAWTs). The LV internal area (LVIA) was measured without including the papillary muscles. Both were recorded in end-systole and end-diastole. The fractional area shortening (FAS) was calculated as [(LVIAED − LVIAES)/LVIAED] × 100 %. Heart rates of mice were controlled above 400/min throughout the echocardiography performance.
Statistical analysis
The results are presented as mean ± SEM. Statistics were calculated using Prism 5.0 (GraphPad Software Inc, San Diego, CA, USA). Linear regression analysis was performed to determine the correlation between two variables. Statistical comparisons for different groups were performed using either the Student’s t test or one-way ANOVA. p values < 0.05 were considered statistically significant.
Results
Characterization of MSCsFluc+GFP+
MSCsFluc+GFP+ were isolated from reporter transgenic mice [Tg(fluc–egfp)] that constitutively express both Fluc and eGFP. Flow cytometry results revealed that MSCsFluc+GFP+ were uniformly positive for MSC markers CD29, CD90, CD44, and negative for CD31, CD45, CD34, SMA, c-Kit (supplemental Fig. 1A). MSCsFluc+GFP+ exhibited fibroblast-like morphology and expressed GFP (supplemental Fig. 1B). The multipotency of MSCsFluc+GFP+ was confirmed by the capacity to differentiate into adipogenic and osteogenic lineages, as assessed by oil red O staining and alizarin red S staining, respectively (supplemental Fig. 1B). Representative bioluminescence images demonstrated a robust linear correlation between the number of MSCsFluc+GFP+ and average Fluc radiance (r 2 = 0.98; supplemental Fig. 1C), indicating that BLI of Fluc was a reliable tool to monitor viable transplanted MSCsFluc+GFP+ quantitatively in vivo.
Hypoxia increased IP6Ks activity and IP7 production
To test the effect of hypoxia injury on the IP6Ks activity and IP7 synthesis, we monitored inositol polyphosphate levels by HPLC (Fig. 1a), which revealed that H/SD injury accumulated large amounts of IP7 (22.7 ± 1.4 % of IP6 vs. 7.3 ± 0.8 % of IP6 in normoxia condition, p < 0.05). Additionally, the formation of IP7 was inhibited by the presence of TNP at 10 μmol/L (12.2 ± 1.6 % of IP6 vs. 22.7 ± 1.4 % of IP6 in H/SD group; Fig. 1b).

IP6Ks activity and IP7 synthesis after hypoxia and serum deprivation (H/SD) injury. a High-performance liquid chromatography (HPLC) profiles of inositol phosphates isolated from MSCsFluc+GFP+ labeled with [2-3H]inositol and stimulated with TNP (0.5, 1, 5, and 10 μmol/L) for 2 h at 37 °C. b Quantitative analysis of a histogram shows IP7 as a percentage of IP6 in MSCs. Data expressed as mean ± SEM. n = 5, *p < 0.05 vs. control (normoxia), # p < 0.05 vs. H/SD group. CPM counts per minute
TNP enhanced the viability of MSCsFluc+GFP+ after H/SD injury
In vitro BLI displayed a remarkable decline of BLI signal intensity in MSCsFluc+GFP+ after H/SD injury compared with normoxic controls (52.8 ± 2.9 × 105 p/s/cm2/sr vs. 165.4 ± 6.8 × 105 p/s/cm2/sr at normal condition, p < 0.05). Moreover, inhibition of IP6Ks with TNP protected the impaired viability of MSCsFluc+GFP+ in a concentration-dependent way after H/SD injury, and TNP pretreatment at 10 μmol/L enhanced the viability maximally (121.7 ± 5.1 × 105 p/s/cm2/sr vs. 52.8 ± 3.0 × 105 p/s/cm2/sr for H/SD, p < 0.05, Fig. 2a). MTT results also confirmed that TNP (10 μmol/L) protected the impaired viability of MSCsFluc+GFP+ after H/SD injury (1.2 ± 0.1 vs. 0.5 ± 0.1 for H/SD, p < 0.05, Fig. 2b). Furthermore, flow cytometry and caspase-3 activity indicated that TNP at 10 μmol/L protected MSCsFluc+GFP+ from apoptosis induced by H/SD injury (15.7 ± 1.0 % vs. 34.3 ± 1.6 %, p < 0.05; Fig. 2c–e).

Effect of TNP on the viability and apoptosis of MSCsFluc+GFP+ after H/SD injury. a In vitro BLI and quantification revealed the survival of MSCsFluc+GFP+ in normal vs. H/SD condition. b MTT assay demonstrated effects of different concentrations of TNP (0.5, 1, 5, and 10 μmol/L) on viability of MSCsFluc+GFP+ after H/SD injury. Representative flow cytometry analyses (c) and quantification (d) for apoptosis of MSCsFluc+GFP+ induced by H/SD. e Histogram illustrated the caspase-3 enzymatic activity in MSCsFluc+GFP+ after H/SD injury. Data expressed as mean ± SEM. n = 5, *p < 0.05 vs. control (normoxia), # p < 0.05 vs. H/SD group
TNP increased Akt activity and growth factors secretions in MSCsFluc+GFP+ after H/SD injury
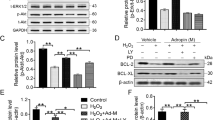
The concentration response effect of TNP on Akt activation was evaluated by phospho-Akt/total Akt. As shown in Fig. 3a, the expression of phospho-Akt was significantly decreased in the H/SD injury group, indicating impaired Akt activation in MSCsFluc+GFP+ during H/SD injury. However, pretreatment of TNP at 10 μmol/L significantly augmented Akt phosphorylation in MSCsFluc+GFP+ after H/SD. Furthermore, TNP administration was also associated with a significant decrease in the activation of pro-apoptotic proteins Bad and a decreased Bax/Bcl-2 ratio in MSCsFluc+GFP+ after H/SD injury (Fig. 3b, c). Moreover, the expressions of Bax were increased after Akt siRNA in all groups, indicating that Akt regulated the expression of Bax (supplemental Fig. 2).

Akt activity and growth factors secretions in MSCsFluc+GFP+ after H/SD injury. Representative blots and semi-quantitative analysis of p-Akt (T308/S47)/Akt (a), Bcl-2/Bax (b), and pBad/Bad (c) expressions in MSCsFluc+GFP+ after H/SD. ELISA assay demonstrated levels of VEGF (d), bFGF (e), IGF-1 (f), and HGF (g) within MSCsFluc+GFP+ supernatants after H/SD. Data expressed as mean ± SEM. n = 5, *p < 0.05 vs. control (normoxia), # p < 0.05 vs. H/SD group
It has been shown that MSCs contribute to cardiac repair and regeneration at least in part by paracrine mechanism. Therefore, we evaluated the effect of TNP on cytokine secretion in MSCsFluc+GFP+ with or without exposure to H/SD. After 6 h H/SD injury, only VEGF (1.1 ± 0.1 × 103 vs. 0.5 ± 0.1 × 103 pg/ml in normoxia condition, p < 0.05) and bFGF (26.6 ± 2.5 vs. 12.1 ± 1.6 pg/ml in normoxia condition, p < 0.05) increased in control groups. Furthermore, TNP administration at 10 μmol/L significantly increased VEGF (3.4 ± 0.3 × 103 vs. 1.3 ± 0.1 × 103 pg/ml, p < 0.05), bFGF (88 ± 5.1 vs. 50 ± 4.8 pg/ml, p < 0.05), IGF-1 (192 ± 4.1 vs. 140 ± 7.8 pg/ml, p < 0.05), and HGF (100 ± 5.7 vs. 49 ± 3.3 pg/ml, p < 0.05) secreted by MSCsFluc+GFP+ after H/SD (Fig. 3d–g).
TNP enhanced the retention of engrafted MSCs
Longitudinal BLI was performed to determine the effect of IP6K inhibition on the retention of MSCs transplanted into infarcted hearts. Representative BLI results in Fig. 4a revealed a progressive decay of BLI signal within 3 weeks after transplantation in MSCs group, which might account for the higher rate of acute-phase mortality. By contrast, TNP facilitated the retention of engrafted MSCsFluc+GFP+ (Fig. 4b). Furthermore, to confirm in vivo BLI results for MSCsFluc+GFP+ survival, histological staining for GFP was performed at 2 or 14 days after transplantation. As shown by the representative immunofluorescence results in Fig. 4c, the numbers of GFP+ cells were not different between groups 2 days after transplantation. By contrast, more GFP+ cells were observed in MSCs + TNP group 2 weeks post-transplantation (366 ± 34.2 vs. 114 ± 17.8 cells/mm2, p < 0.05, Fig. 4c, d). These data suggested that IP6K inhibition with TNP promoted the retention of engrafted MSCs.

Tracking the survival of transplanted MSCsFluc+GFP+. a Longitudinal BLI spatiotemporally tracked MSCsFluc+GFP+ survival in MSCs group (top row, n = 10) and MSCs + TNP group (bottom row, n = 10). b Quantitative analysis of Fluc optical signals on fixed regions of interest (ROI). c Representative immunofluorescence images of triple staining for GFP (green fluorescent), cTn I (red fluorescent) and DAPI (blue fluorescence) at 2 and 14 days after transplantation (Scale bars 50 μmol/L). (d) Quantitative analysis of GFP-positive cells in MSCs group and MSCs + TNP group on POD 2 and 14. Data expressed as mean ± SEM. n = 5, *p < 0.05 vs. MSCs group, # p < 0.05 vs. POD 2
TNP facilitated MSC-mediated pro-angiogenic and anti-apoptotic effects
To investigate the effects of TNP on the acute protective function of engrafted MSCs, we firstly evaluated the apoptosis index in the heart’s border zone around infarcted area at 48 h after ligation of anterior descending artery. Moreover, we also evaluated the revascularization in the border zone around infarcted area 4 weeks post-operation. Immunofluorescence staining revealed that the number of CD31-positive cells in the MI group was no significantly different from that in the sham group (199.7 ± 35.5 vs. 210.0 ± 20.0 cell/mm2, p > 0.05, Fig. 5a, c). Transplantation of MSCs that were pretreated with TNP significantly increased the CD31+ cells compared to MSCs group (450.0 ± 10.0 vs. 330.0 ± 20.0 cell/mm2 p < 0.001, Fig. 5a, c). Consistently, the number of capillaries, as evaluated by immunohistochemistry of CD31, was elevated in MSCs + TNP group (17.0 ± 1.0 vs. 10.7 ± 1.5, p = 0.004, Fig. 5b, d). TUNEL assay analysis revealed more apoptotic cells in the heart of MI group compared to sham group (31.6 ± 2.1 vs. 9.2 ± 0.5 %, p<0.05, Fig. 6a, b). Moreover, transplantation of MSCs pretreated with TNP significantly decreased the apoptosis of cells in the heart compared to the MSCs without TNP administration (18.5 ± 0.9 vs. 27.3 ± 0.8 %, p < 0.05).

TNP pretreated MSC transplantation promoted angiogenesis. a Representative confocal microscopic images of triple immunofluorescence staining for CD31 (red fluorescent), cTnI (green fluorescent) and DAPI (blue fluorescent) on POD28. b Capillaries in the infarct border zone were determined by immunohistochemical staining for CD31-positive cells (red arrows, scale bars 50 μmol/L). Quantitative analysis of the CD31-positive cells (c) and capillaries in the infarct border zone (d) demonstrated TNP pretreated MSC transplantation promoted angiogenesis. Error bars represent mean ± SD. n = 5, *p < 0.005 vs. Sham, # p < 0.05 vs. MI

TNP pretreated MSC transplantation decreased apoptosis in the heart. a Confocal microscopy of TUNEL staining for cell apoptosis in border zone around infarcted area 48 h after LAD ligation in mice hearts. Apoptotic nuclei were identified as TUNEL positive (green fluorescent). Myocardium was stained using a monoclonal antibody against cTnI (red fluorescent) and total nuclei by DAPI counterstaining (blue fluorescent). Scale bar represents 50 μmol/L. b TUNEL-positive apoptotic cells were quantified by apoptotic index (AI), which was defined as the percentage of apoptotic cells. n = 5, *p < 0.05 vs. sham, # p < 0.05 vs. MI
MSCs pretreated with TNP reduced fibrosis and preserved heart functional recovery after MI
To study the effects of TNP pretreatment on the therapeutic efficiency of engrafted MSCs for MI, we performed fibrosis and function analysis. As illustrated in Fig. 7a and b, severe fibrosis was observed in the hearts of MI group and MSCs group. Conversely, transplantation of MSCs pretreated with TNP decreased the area of fibrosis significantly compared with MI group (Fig. 7c). Serial echocardiographic analysis indicated that the baseline parameters were similar in all groups. The LV dimensions (LVEDD, LVESD) were increased after MI. However, the LV dimensions were decreased in MSCs + TNP group compared with MI and MSCs group (Fig. 7d, e). Transplantation of MSCs with TNP also manifested a trend towards improvement of cardiac performance over the 4 weeks after MI (Fig. 7f, g, h, i).

Evaluation of fibrosis and heart function. a Representative fast green/sirius red staining revealed left ventricular fibrosis 4 weeks after MI (magnification 4×). b Histological examination of fibrosis by fast green/sirius red staining in the border zone. Green indicates viable myocardium, while red indicates fibrosis due to infarction damage. Scale bar represents 50 μmol/L. c Quantitative analysis of the fibrotic area. Histograms illustrated heart function parameters: LVESV (d), LVEDV (e), LVEF (f), and LVFS (g). n = 5, *p < 0.05 vs. MI
Discussion
In the present study, we found that IP6K inhibition exerted a protective effect on the survival of MSCs under hypoxia, which was associated with decreased production of IP7 and increased Akt activity. Intra-myocardial injection of bone marrow-derived MSCs pretreated with IP6ks inhibitor exerted even greater cardioprotection against ischemic injury. Overall, this study demonstrates that transplantation of MSCs with IP6K inhibition during acute phase of MI is an effective strategy for cardiac protection.
Although both experimental and clinical studies of MSC-based therapy for MI have reached an exciting and promising stage, previous studies revealed only marginal improvements in cardiac function after the transplantation of MSCs into infarcted hearts [26]. Poor viability of donor cells is thought to be a major limitation for MSCs’ clinical application. Our previous studies have demonstrated a high level of MSCs death between day 3 and 7 after implantation [34]. In the present study, we tracked the engrafted MSCs by BLI and also found similar acute cell death within 1 week after transplantation into the infarcted heart. Meanwhile, transplantation with MSCs alone could not prevent cardiomyocytes death and improve cardiac function significantly, which was consistent with the results of Yang et al. [31, 32]. Many factors are believed to contribute to the high losses of MSCs after transplantation. The noxious milieu in ischemic myocardium that lacks nutrients and oxygen, coupled with enhanced inflammatory response, may play important roles here [5, 12]. Moreover, donor MSCs seem more sensitive to hypoxic and inflammatory injury due to the stressful procedure of cell preparation and transplantation [21]. Therefore, reinforcement of the viability of MSCs in hypoxic conditions after transplantation is crucial for improving the efficiency of cell therapy.
Akt, a PH domain containing serine/threonine kinase, regulates survival signal, growth factor production, and protein synthesis through the phosphorylation of multiple substrates [7]. The Bcl-2 family, regulated by Akt activity, makes up the key regulators of apoptosis [1, 25]. Moreover, previous studies [15, 18, 29] also have proved that increased Akt activation resulted in decreased expression of Bax and increased expression of Bcl-2, suggesting that Akt activation was negatively correlated with the expressions of pro-apoptosis proteins, Bax. In the present study, we found that the phosphorylation of Akt was decreased by hypoxia injury, associated with an increased Bax/Bcl-2 ratio. However, these effects were abolished by TNP, indicating that IP6Ks inhibition protected the impaired activation of Akt induced by H/SD injury. Therefore, protection of impaired Akt activation is seen as an attractive therapeutic target to optimize MSCs therapy for ischemic heart disease. Various strategies have been adopted to increase the Akt activation of engrafted MSCs. Mangi et al. [15] found that the genetically modified MSCs (Akt-MSCs) exhibited significantly enhanced intramyocardial retention and exerted a potent myocardial protection effect by paracrine mechanisms. Haider et al. [11] also revealed that increasing Akt activation by over-expressing insulin-like growth factor (IGF)-1 promoted MSCs survival and enhanced their therapeutic efficacy. However, the potential tumorigenicity risk and higher manufacturing cost might hamper genetically engineered cell therapy for the clinic. An alternative approach of gene delivery is the pretreatment with small molecules to up-regulate donor cells Akt activity.
Inositol phosphates (IPs) are a group of signaling molecules that are widely distributed in all eukaryotic cells [2]. IP6 can be further metabolized to generate 5-diphosphoinositol pentakisphosphate (IP7) by a family of three IP6 kinases (IP6Ks) [24]. Although the physiological effects of IPs remain poorly characterized, IP7 appears to be involved in cell apoptosis [13]. In the present study, we found increased apoptotic MSCs with significantly enhanced IP7 generation after hypoxia injury. Moreover, the IP6K inhibition by TNP was also associated with decreased apoptosis, increased viability and paracrine effect of MSCs, indicating that IP7 may be involved in the hypoxia injury of MSCs.
IP7 could inhibit Akt signaling by binding at the PH domain of Akt to block phosphorylation both in vitro and in vivo [6]. In the present study, we demonstrated that blocking IP6Ks by TNP decreased IP7 synthesis and increased phosphorylation of Akt at T308 and S473 in MSCs, indicating the down-regulation of IP7 expression by IP6K inhibition enhanced the activation of Akt in MSCs. Furthermore, we documented that blocking IP6Ks using a pharmacological inhibitor TNP improved the survival of MSCs in the ischemic heart. Brunner et al. [4] found boosted migration of MSCs from bone marrow to peripheral blood and heart reduced apoptosis of cardiomyocytes and improved cardiac function significantly. Our research also proved that transplantation with TNP pretreated MSCs was effective at preventing apoptosis of cells in the heart and myocardial fibrosis, leading to improved LV function.
Although there is controversy on the precise mechanisms underlying these therapeutic effects associated with engrafted stem cells [14, 19], a growing body of evidence supports the hypothesis that paracrine mechanisms play a more essential role in stem cell-mediated tissue protection instead of cardiac differentiation [10]. Moreover, many bioactive factors secreted by MSCs, such as VEGF, bFGF, HGF, and IGF-1, promote neovascularization and inhibit host cardiomyocyte death [17]. However, the effect of hypoxia injury on the paracrine effects of MSCs is still not clear. In the current study, we found that the secretions of VEGF and bFGF in MSCs under hypoxic condition were increased compared with normal groups. However, it seems a contradiction that hypoxia significantly decreased IGF-1 and HGF in MSCs. We presumed that the low-grade augmentation of the VEGF and bFGF secretions maybe a compensatory response of MSCs to hypoxic injury [28, 30]. Meanwhile, we found that IP6Ks inhibition significantly increased VEGF, bFGF, IGF-1, and HGF secreted by MSCs after hypoxia injury. Taken together, our results demonstrated that TNP administration protected the impaired paracrine effects of MSCs under hypoxia.
Although our study bears clinical relevance, there are some limitations. First, physiological functions of IP7 have not been extensively characterized. Further studies defining the exact mechanism(s) are needed. Second, the H/SD model is an artificial experimental model that cannot fully simulate the in vivo ischemic and inflammatory environment.
In conclusion, the current study confirmed that increasing IP7 formation by IP6Ks can lead to impaired viability and paracrine effect of MSCs engrafted into ischemic heart. However, inhibition of IP6Ks has the potential to enhance functional survival of engrafted MSCs via activation of Akt signaling, which was associated with preserved cardiac function (Fig. 8). Therefore, our results indicate that IP6Ks and IP7 may be promising novel modification targets for augmenting MSC therapeutic efficacy.

Schematic depicting IP6Ks/IP7 regulation of MSCs for myocardial infarction. Hypoxia stimulates IP6Ks activation and IP7 formation in MSCs, leading to pronounced inhibition of Akt with impaired survival and paracrine effects. Inhibition of IP6Ks has the potential to enhance the survival and paracrine effects of engrafted MSCs via activation of Akt signaling. Thus, IP6Ks may be a potential target for optimizing MSC therapy for MI
References
Belkhiri A, Dar AA, Zaika A, Kelley M, El-Rifai W (2008) t-Darpp promotes cancer cell survival by up-regulation of Bcl2 through Akt-dependent mechanism. Cancer Res 68:395–403. doi:10.1158/0008-5472.CAN-07-1580
Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, Molina H, Pandey A, Werner JK Jr, Juluri KR, Xu Y, Prestwich GD, Parang K, Snyder SH (2007) Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci USA 104:15305–15310. doi:10.1073/pnas.0707338104
Braunwald E, Bristow MR (2000) Congestive heart failure: fifty years of progress. Circulation 102:IV14–IV23
Brunner S, Theiss HD, Leiss M, Grabmaier U, Grabmeier J, Huber B, Vallaster M, Clevert DA, Sauter M, Kandolf R, Rimmbach C, David R, Klingel K, Franz WM (2013) Enhanced stem cell migration mediated by VCAM-1/VLA-4 interaction improves cardiac function in virus-induced dilated cardiomyopathy. Basic Res Cardiol 108:388. doi:10.1007/s00395-013-0388-3
Cao F, Lin S, Xie X, Ray P, Patel M, Zhang X, Drukker M, Dylla SJ, Connolly AJ, Chen X, Weissman IL, Gambhir SS, Wu JC (2006) In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation 113:1005–1014. doi:10.1161/CIRCULATIONAHA.105.588954
Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH (2010) Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 143:897–910. doi:10.1016/j.cell.2010.11.032
Chan TO, Rittenhouse SE, Tsichlis PN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68:965–1014. doi:10.1146/annurev.biochem.68.1.965
Dec GW, Fuster V (1994) Idiopathic dilated cardiomyopathy. N Engl J Med 331:1564–1575. doi:10.1056/NEJM199412083312307
Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ (2010) A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107:1445–1453. doi:10.1161/CIRCRESAHA.110.223925
Gnecchi M, Zhang Z, Ni A, Dzau VJ (2008) Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 103:1204–1219. doi:10.1161/CIRCRESAHA.108.176826
Haider H, Jiang S, Idris NM, Ashraf M (2008) IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ Res 103:1300–1308. doi:10.1161/CIRCRESAHA.108.186742
Li Z, Wu JC, Sheikh AY, Kraft D, Cao F, Xie X, Patel M, Gambhir SS, Robbins RC, Cooke JP, Wu JC (2007) Differentiation, survival, and function of embryonic stem cell derived endothelial cells for ischemic heart disease. Circulation 116:I46–I54. doi:10.1161/CIRCULATIONAHA.106.680561
Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH (2003) Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell 114:559–572
Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, Sano M, Takahashi T, Hori S, Abe H, Hata J, Umezawa A, Ogawa S (1999) Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest 103:697–705. doi:10.1172/JCI5298
Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, Dzau VJ (2003) Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med 9:1195–1201. doi:10.1038/nm912
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. doi:10.1016/j.cell.2007.06.009
Markel TA, Wang Y, Herrmann JL, Crisostomo PR, Wang M, Novotny NM, Herring CM, Tan J, Lahm T, Meldrum DR (2008) VEGF is critical for stem cell-mediated cardioprotection and a crucial paracrine factor for defining the age threshold in adult and neonatal stem cell function. Am J Physiol Heart Circ Physiol 295:H2308–H2314. doi:10.1152/ajpheart.00565.2008
Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M (1999) Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem 73:2037–2046
Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P (2001) Bone marrow cells regenerate infarcted myocardium. Nature 410:701–705. doi:10.1038/35070587
Padmanabhan U, Dollins DE, Fridy PC, York JD, Downes CP (2009) Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem 284:10571–10582. doi:10.1074/jbc.M900752200
Reinecke H, Murry CE (2002) Taking the death toll after cardiomyocyte grafting: a reminder of the importance of quantitative biology. J Mol Cell Cardiol 34:251–253. doi:10.1006/jmcc.2001.1494
Reinecke H, Zhang M, Bartosek T, Murry CE (1999) Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation 100:193–202
Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH (1999) Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol CB 9:1323–1326
Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH (2001) Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem 276:39179–39185. doi:10.1074/jbc.M106842200
Sheikh AM, Malik M, Wen G, Chauhan A, Chauhan V, Gong CX, Liu F, Brown WT, Li X (2010) BDNF-Akt-Bcl2 antiapoptotic signaling pathway is compromised in the brain of autistic subjects. J Neurosci Res 88:2641–2647. doi:10.1002/jnr.22416
Spinelli SV, Rodriguez JV, Quintana AB, Mediavilla MG, Guibert EE (2002) Engraftment and function of intrasplenically transplanted cold stored rat hepatocytes. Cell Transplant 11:161–168
Sun D, Huang J, Zhang Z, Gao H, Li J, Shen M, Cao F, Wang H (2012) Luteolin limits infarct size and improves cardiac function after myocardium ischemia/reperfusion injury in diabetic rats. PLoS One 7:e33491. doi:10.1371/journal.pone.0033491
Tong Q, Zheng L, Lin L, Li B, Wang D, Huang C, Li D (2006) VEGF is upregulated by hypoxia-induced mitogenic factor via the PI-3 K/Akt-NF-kappaB signaling pathway. Respir Res 7:37. doi:10.1186/1465-9921-7-37
Yang L, Guo W, Zhang Q, Li H, Liu X, Yang Y, Zuo J, Liu W (2011) Crosstalk between Raf/MEK/ERK and PI3 K/AKT in suppression of Bax conformational change by Grp75 under glucose deprivation conditions. J Mol Biol 414:654–666. doi:10.1016/j.jmb.2011.09.009
Yang XM, Wang YS, Zhang J, Li Y, Xu JF, Zhu J, Zhao W, Chu DK, Wiedemann P (2009) Role of PI3K/Akt and MEK/ERK in mediating hypoxia-induced expression of HIF-1alpha and VEGF in laser-induced rat choroidal neovascularization. Invest Ophthalmol Vis Sci 50:1873–1879. doi:10.1167/iovs.08-2591
Yang YJ, Qian HY, Huang J, Geng YJ, Gao RL, Dou KF, Yang GS, Li JJ, Shen R, He ZX, Lu MJ, Zhao SH (2008) Atorvastatin treatment improves survival and effects of implanted mesenchymal stem cells in post-infarct swine hearts. Eur Heart J 29:1578–1590. doi:10.1093/eurheartj/ehn167
Yang YJ, Qian HY, Huang J, Li JJ, Gao RL, Dou KF, Yang GS, Willerson JT, Geng YJ (2009) Combined therapy with simvastatin and bone marrow-derived mesenchymal stem cells increases benefits in infarcted swine hearts. Arterioscler Thromb Vasc Biol 29:2076–2082. doi:10.1161/ATVBAHA.109.189662
Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE (2001) Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol 33:907–921. doi:10.1006/jmcc.2001.1367
Zhang Z, Li S, Cui M, Gao X, Sun D, Qin X, Narsinh K, Li C, Jia H, Han Y, Wang H, Cao F (2013) Rosuvastatin enhances the therapeutic efficacy of adipose-derived mesenchymal stem cells for myocardial infarction via PI3K/Akt and MEK/ERK pathways. Basic Res Cardiol 108:333. doi:10.1007/s00395-013-0333-5
Zhang Z, Li W, Sun D, Zhao L, Zhang R, Wang Y, Zhou X, Wang H, Cao F (2011) Toll-like receptor 4 signaling in dysfunction of cardiac microvascular endothelial cells under hypoxia/reoxygenation. Inflamm Res 60:37–45. doi:10.1007/s00011-010-0232-2
Acknowledgments
This work was supported by National Nature Science Foundation of China (No. 81325009, 81270168, 81090274, 81227901), National Basic Research Program of China (2012CB518101), Program for Changjiang Scholars and Innovative Research Team in University (IRT1053), China’s Ministry of Science and Technology 863 Program (2012AA02A603).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Z. Zhang, D. Liang, and X. Gao contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Z., Liang, D., Gao, X. et al. Selective inhibition of inositol hexakisphosphate kinases (IP6Ks) enhances mesenchymal stem cell engraftment and improves therapeutic efficacy for myocardial infarction. Basic Res Cardiol 109, 417 (2014). https://doi.org/10.1007/s00395-014-0417-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-014-0417-x