
Abstract
Liver X receptor (LXR)-α and -β play a major role in lipid and glucose homeostasis. Their expression and function in the heart is not well characterized. Our aim was to describe the expression of LXRs in the murine heart, and to determine effects of cardiac LXR activation on target gene expression, lipid homeostasis and ischemia. Both LXRα and -β were expressed in heart tissues, HL-1 cells and isolated cardiomyocytes as determined by qRT-PCR. Elevated cardiac expression of LXR target genes and LXRβ was observed 24 h after in vivo permanent coronary artery ligation. The synthetic LXR agonist GW3965 induced mRNA expression of the LXR target genes in HL-1 cells and isolated cardiomyocytes. This was associated with a buildup of intracellular triglycerides and expanding lipid droplets as quantified by confocal microscopy. Mice injected with GW3965 had cardiac LXR activation as judged by increased target gene expression and lipid droplet accumulation. GW3965 in vivo and in vitro increased expression of genes inducing triglyceride synthesis, and altered expression of lipid droplet-binding protein genes. GW3965 protected HL-1 cells against hypoxia-reoxygenation induced apoptosis. LXR activation by GW3965 in vivo prior to heart isolation and perfusion with induced global ischemia and reperfusion improved left ventricular contractile function and decreased infarct size. In conclusion, LXRs are expressed in the murine heart in the basal state, and are activated by myocardial infarction. Activation of LXR by the synthetic agonist GW3965 is associated with intracardiac accumulation of lipid droplets and protection against myocardial ischemia–reperfusion injury.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Liver X receptors (LXRs) are nuclear receptors that regulate genes involved in lipid and glucose metabolism [7]. The two LXR isoforms LXRα and LXRβ form heterodimers with retinoid X receptor (RXR), which bind to genomic LXR response elements [3]. Oxidized metabolites of cholesterol are natural ligands that bind and activate LXRs [23]. Modulation of dual LXRα/β activity can be achieved by using synthetic compounds such as the full agonist T0901317 and partial but more specific agonist GW3965 [33, 42]. LXR ligand-binding results in distinct conformational changes which may increase genomic binding of heterodimers as well as shift the recruitment of corepressor and coactivator complexes to regulate transcription [39, 56]. The two LXR isoforms show distinct expression patterns; LXRα has its highest expression in particular tissues such as the liver, adipose tissue, small intestine, spleen, macrophages, and kidney; whereas LXRβ is ubiquitously expressed.
LXRs regulate expression of genes involved in lipid and carbohydrate metabolism in liver, adipose tissue, as well as skeletal muscle [7, 21, 25]. Their role in hepatic de novo lipogenesis is well characterized [42]: LXRs act indirectly by targeting the sterol-regulatory element-binding protein 1c (SREBP-1c) gene, a master regulator of lipogenic genes like acetyl CoA carboxylase and fatty acid synthase [9, 52]; and directly by activating transcription of other genes like the stearoyl-coenzyme A desaturase 1 (SCD1) gene [11]. The SCD1 protein introduces double bonds in saturated fatty acids (16:0 and 18:0) to make monosaturates (16:1 and 18:1). LXR also promotes reverse cholesterol transport, from the periphery to the liver for excretion, by targeting ATP-binding cassette (ABC) transporter genes like ABCA1 [8, 46].
Fatty acids are the major energy substrate for the heart under most physiological conditions, and are obtained either by uptake from the circulation or hydrolysis of intracellular triglycerides [20, 51]. Ligation of fatty acids to CoA by CoA synthetase 3 (ACSL3) activates them for beta-oxidation in the mitochondria to support ATP production or, alternatively, incorporation into triglycerides by glycerol-3-phosphate acyltransferase (GPAM, also known as GPAT1) and diacylglycerol O-acyltransferase 2 (DGAT2). Triglycerides accumulate together with other hydrophobic lipids in the core of cytosolic lipid droplets, surrounded by a phospholipid monolayer and the perilipin scaffolding proteins (PLIN1–PLIN5) [40]. When triglycerides stored in lipid droplets are to be used as energy substrate, they are hydrolyzed involving abhydrolase domain containing 5 (Abhd5, also known as CGI-58) and phospholipase domain containing 2 (PNPLA2, also known as ATGL) to generate glycerol and fatty acids [29].
The role of lipids in the heart is controversial; excess of lipid compounds in the cytosol such as fatty acids, triglycerides, ceramides, and diglycerides may lead to lipotoxicity and loss of cardiac function, whereas their compartmentalization into lipid droplets has been associated with protective effects [4, 5, 10, 45, 53, 55]. Cardiomyocytes accumulate lipid droplets during fasting and exercise, or under pathological conditions such as ischemia, diabetes or obesity [2, 20, 24, 31, 49]. In myocardial ischemia, suppression of fatty acid uptake and oxidation increases myocardial glucose utilization [6, 26]. Thus, in the regionally ischemic heart lipid droplets accumulate mainly in the periphery of the area at risk [24, 48]. In isolated cardiomyocytes exposed to anoxia and reoxygenation, cell death was attenuated in cells containing lipid droplets, indicating cardioprotective effects [1]. The heart expresses several PLIN genes, and genetic ablation of PLIN5 lead to reduction of cardiac lipid droplet formation and increased production of reactive oxygen species [28]. PLIN5 may also protect mitochondria in cardiomyocytes against excessive fatty acid exposure during stress [53].
LXRs have been implicated to play beneficial roles in cellular responses to ischemia. A single dose of LXR ligand administered post-injury dramatically reduced brain damage by reducing cell death in a rodent hippocampus model of acute brain ischemia [43]. In a rodent model of splanchnic ischemia, LXR activation reduced mortality, attenuated the inflammatory response and increased cell survival [15]. In the heart, LXRs may regulate cardiac hypertrophy [57]; however, their role in myocardial ischemia and reperfusion has not been evaluated [44]. We hypothesized that LXRs are expressed in the heart, and that ligand stimulation of LXR would lead to cardiac lipid droplet accumulation and protection against ischemic injury.
Methods
Experimental animals
C57BL/6 male mice weighting 25 ± 1 g, with conventional microbiological status, were used (NOVA-SCB, Nittedal, Norway). All animals were acclimatized for 14 days before experiments in a controlled environment of 12:12-h light/dark photoperiod; 23 °C temperature; 55–60 % humidity; and chow (RM3; Special Diets Service, Witham GB) and tap water ad libitum. The experiments were approved by the Norwegian Animal Health Authority. Mice were killed by neck dislocation or by injecting i.p. with sodium pentobarbital (50 mg/kg; Sigma-Aldrich, St. Louis, USA) with addition of heparin (500 IU; Leo Pharma, Oslo, Norway) when used for heart perfusions.
Gene expression analyses
To investigate basal mRNA expression of LXRs in tissues (n = 3), liver, heart, and m. gastrocnemius were harvested immediately in RNAlater (Qiagen, Hilden, Germany). The hearts were further divided into left and right ventricular walls, ventricular septum and atria (left and right together) before RNA extraction and gene expression analyses using qRT-PCR. For comparison, gene expression in cardiomyocytes, cardiofibroblasts, and HL-1 cells as described below was investigated. Primer sequences and additional methodological details are provided online (Supplementary Table 2; Detailed Methods Description in Supplementary Material).
In vivo myocardial ischemia
To investigate if myocardial ischemia would evoke endogenous activation of LXR signaling, in vivo myocardial infarction was induced by permanent occlusion of the left coronary artery (n = 9) or sham operation (n = 9) with procedures described elsewhere [16]. 24 h later the mice were killed for RNA extraction and qRT-PCR gene expression analyses using an ABI Prism 7900 instrument (Life Technologies, Carlsbad, USA).
Isolated cardiomyocytes
To evaluate if LXR target genes were activated by GW3965, primary cardiomyocytes were isolated following the procedure described by O’Connell et al. [36]. Isolated cardiomyocytes were resuspended in medium and left to attach for 4 h at 37 °C and 2 % CO2. GW3965 was dissolved in DMSO (Sigma-Aldrich) to make a 1 mM stock solution. To activate LXR, isolated cardiomyocytes were changed to fresh medium supplemented with 0.1 % DMSO ± 1 μM GW3965 after the attachment period, and incubated for 20 h before harvest. Non-cardiomyocytes present in the supernatant from the first low-speed centrifugation were plated on non-coated six-well plates incubated for 4 h at 37 °C and 5 % CO2. Three individual experiments were repeated in duplicates. Additional information is provided online (Detailed Methods Description, Supplementary Material).
HL-1 cells
HL-1 cells were used to study the dose–response effect of GW3965 on LXR target gene expression, and to evaluate if LXR agonism led to lipid droplet formation. HL-1 cells were cultured as previously described [12]. Cells were seeded on gelatine/fibronectin-coated plastic cluster plates or flasks (BD Biosciences, Franklin Lakes, USA) at a density of 5 × 105 cells/well and cultured in Claycomb medium with supplements as described previously [12] at 37 °C and 5 % CO2. The synthetic LXR agonist GW3965 was given at 0.001–5 μM ligand for 18 h to cells seeded in six-well plates, and cells were harvested, RNA extracted and qRT-PCR performed (online supplement). Maximum effect of GW3965 on target gene mRNA expression was observed at concentrations 0.1–1 μM with a small inhibitory effect at the higher concentration (5 μM). 1 μM GW3965 was used in all succeeding experiments. The effect of GW3965 incubation on lipid accumulation was tested in HL-1 cells incubated for 48 h in Claycomb medium supplemented with delipidized FBS (Lonza, Basel, Switzerland) in the presence of 200 μM low endotoxin fatty acid free BSA ± 500 μM conjugated sodium oleate (Nu-Chek-Prep, Elysian, USA), and 0.1 % DMSO ± 1 μM GW3965 [19]. Intracellular lipid droplets were evaluated in cells cultured on 8-well Lab-Tek II chamber slides (Thermo Fisher Scientific, Waltham, USA) by confocal microscopy using ImageJ (see Detailed Methods, Supplemental Material) after staining with 10 μM Bodipy 493/503 (Life Technologies) and 10 μM Hoechst 33342 (Sigma-Aldrich). The total intracellular triglyceride content in HL-1 cells was analyzed (n = 9) colorimetrically using the Triglycerides Enzymatique Kit (BioMérieux, Marcy l’Etoile, France) in homogenates obtained by growing cells for 48 h in T-75 flasks in the media above, and then scraping them into PBS, pelleting them by centrifugation, and finally lysing them in RIPA buffer (Thermo Fisher Scientific). Hypoxia-reoxygenation experiments were performed to investigate expression of apoptosis proteins after LXR ligand stimulation. Confluent HL-1 cells were stimulated with 1 μM GW3965 with 500 μM sodium oleate or vehicle alone for 18 h. Thereafter, cells were kept in a hypoxia chamber glove box (model #856-HYPO; Plas-Labs, Lansing, USA) under normoxic (5 % CO2, 20 % O2, 75 % N2) or hypoxic (5 % CO2, 0.5 % O2, 94.5 % N2) conditions at 37 °C for 2 h, followed by 8 h reoxygenation in a conventional normoxic cell incubator. Cells were harvested for protein extraction. Experiments in HL-1 cells were repeated three times in triplicates.
Lipid analyses of cardiac tissue
To investigate if LXR stimulation led to altered lipid profile and in vivo lipid droplet accumulation, mice were fasted overnight and injected i.p. three times at 8 h intervals with GW3965 (20 mg/kg) (n = 6) or vehicle alone (PBS + 10 % DMSO) (n = 6). The mice were killed by neck dislocation 8 h after the last injection. The ventricles were dissected out and the apexes excised from them, frozen in liquid N2 and stored under Ar gas at −70 °C before triglyceride extraction and fatty acid profile analysis by gas liquid chromatography with flame ionization detection. The remaining part of the ventricles were fixed for 24 h at room temperature in 4 % paraformaldehyde and dehydrated for 24 h in 20 % sucrose before they were embedded and frozen in OCT (Sakura Finetek, Alphen aan den Rijn, The Netherlands). Transverse ventricular sections of 7 μm thicknesses were prepared using a cryostat (CM3050S; Leica Microsystems, Wetzlar, Germany) at −17 °C on positively charged Superfrost slides (Thermo Fisher Scientific), which were stored at −70 °C until analysis. Another investigator, blinded to the protocol, imaged and analyzed the cryosections. Two cryosections from each mouse were thawed and rehydrated, incubated with 10 μM Bodipy 493/503 (Life Technologies) and 10 μM Hoechst 33342 at 37 °C for 15 min, and mounted using Dako mounting medium (Dako, Glostrup, Denmark). Three confocal images were acquired from each cryosection using an Olympus IX81 confocal microscope (Olympus, Tokyo, Japan). Images were analyzed using the Image J software to determine the number, size and area of lipid droplets. Additional information is provided online (Detailed Methods Description, Supplementary Material).
Western blot
Western blotting was used to investigate the apoptosis protein (Bcl-2, Bax, Caspase 3) expressions in hypoxia/reoxygenation HL-1 cells. After hypoxia/reoxygenation, cells were lysed in the RIPA buffer (pH 7.5 50 mM Tris–HCl, 150 mM NaCl, 1 % NP-40, 1.25 % deoxycholic acid, 1 mM EDTA, protease and phosphatase inhibitors (Thermo Scientific Pierce). Protein concentrations were measured using BCA Protein Assay Kit (Thermo Scientific Pierce); 10-μg total protein was loaded in each lane and separated by SDS-PAGE using a 10 % Tris gel with Tris running buffer at 100 V for 2 h; electroblotted onto nitrocellulose membranes (Amersham Pharmacia Biotech). After 1 h’s blocking in 5 % w/v skimmed milk TBS 0.1 % Tween-20, the membrane was probed with primary antibodies against Bcl-2 (N-19; Santa Cruz Biotechnology, Inc., California, USA), Bax (N-20; Santa Cruz Biotechnology, Inc.), cleaved Caspase 3 (Asp 175; Cell Signaling Technology) in 5 % w/v BSA TBS 0.1 % Tween-20, after 3 times wash with TBS 0.1 % Tween-20, the secondary antibody was added. The signal was detected with enhanced chemiluminescence (ECL Plus, Thermo Scientific Pierce) and autoradiography using X-ray film. Reversible Ponceau staining was used as a loading control.
Isolated heart perfusion
To investigate if LXR agonism protected against myocardial ischemia, hearts were isolated and perfused in a Langendorff model as described previously [41]. A single i.p. injection of GW3965 (20 mg/kg) (n = 7) or vehicle alone (PBS + 10 % DMSO) (n = 7) was given to overnight fasted mice, which were killed 6 h later. Hearts were isolated, cannulated and perfused with a stabilization period of 20 min. A polyethylene balloon was inserted into the left ventricle via the left atrium for isovolumetric pressure registration. Global ischemia was achieved by clamping the inflow tubing for 40 min, followed by reperfusion for 60 min. After this, transverse sections of the hearts were stained using triphenyl tetrazolium chloride (TTC) and infarct area was evaluated blindly. Additional information is provided online (Detailed Methods Description, Supplementary Material).
Statistical analyses
All data are expressed as mean + SEM. Statistical analyses were performed using Prism (GraphPad Prism software, La Jolla, USA). Pairwise comparisons were tested statistically with unpaired/paired t tests (Mann–Whitney test was used in Fig. 2). One-way ANOVA was used in experiments with more than two groups, and two-way ANOVA in designs with two independent variables. Posttests were done using Dunnett’s multiple comparison test. P values <0.05 were considered statistically significant.
Results
Cardiac expression of LXRs
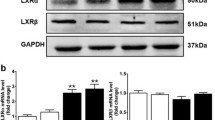
Both LXRα and -β were expressed in the murine heart as evaluated by qRT-PCR (Fig. 1a). The relative gene expression of LXRβ was 5–6 times higher than LXRα. The LXR expression was similar in the left ventricle, the right ventricle, and the ventricular septum. In the atria, mRNA levels of both LXR isoforms were threefold higher than in the ventricle. Ventricular LXRα and -β mRNA levels were approximately 1.5- to 3-fold higher than in skeletal muscle, respectively. On the other hand, left ventricular LXRα and -β mRNA levels were 22- and 3-fold lower, respectively, than in the liver, which is the tissue of highest LXRα expression.

Gene expression of Liver X receptors (LXRα and -β) in the heart. The level of mRNA was determined by qRT-PCR and normalized to GAPDH. a LXRα and -β mRNA expression in left ventricle (LV), right ventricle (RV), septum, atria, skeletal muscle (SkM), and liver from non-fasting adult C57BL/6 male mice (n = 3). b LXRα and -β mRNA expression in murine cells (n = 9): HL-1 cardiomyocyte cell line, isolated primary cardiomyocytes (CM), a mixed cell population containing cardiofibroblasts and endothelial cells (Non-CM). Data are given as mean + SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus LV or CM
Expression of LXRs was also investigated in cells isolated from adult mouse hearts, digested by collagenase and fractionated based on their buoyant properties. LXRα and -β mRNA expression in the cardiomyocyte fraction was comparable with the expression in ventricular tissue (Fig. 1b). A pooled fraction of the remaining non-cardiomyocyte cells displayed LXRα and -β expression levels 10- to 15-fold higher than the cardiomyocytes. This fraction consisted mainly of endothelial cells and fibroblasts, judged by their rapid adherence to the growth surface and the expression of the fibroblast marker gene vimentin and the endothelial marker cadherin (data not shown). In addition, the HL-1 cardiomyocyte cell line expressed both LXRα and -β. Thus, both primary and HL-1 cardiomyocytes are suited for exploring cellular effects of LXR activation.
Myocardial infarction is associated with increased cardiac expression of LXR target genes and LXRβ
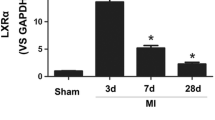
Myocardial infarction was induced by permanent ligation of the left anterior descending coronary artery in mice. Gene expression levels in the hearts were compared after 24 h to those of sham-operated animals. LXR target genes ABCA1, SREBP-1c and SCD1 mRNA levels were increased post-infarction by 22, 44 and 56 %, respectively (Fig. 2). LXRβ was increased by 28 % in infarcted hearts, whereas post-infarction LXRα levels were unchanged (Fig. 2).

Myocardial infarction increases levels of selected LXR target genes and LXRβ expression in the heart. Mice were subjected to either myocardial infarction induced by ligation of the left anterior descending coronary artery (infarction n = 9) or sham operation (sham n = 9). Hearts were harvested 24 h later, mRNA was extracted from the apex, and gene expression levels of Stearoyl-Coenzyme A desaturase 1c (SREBP-1c), ATP-binding cassette transporter A1 (ABCA1), stearyl CoA desaturase 1 (SCD1), LXRα, and LXRβ were determined by qRT-PCR and normalized to GAPDH. Bars represent group means + SEM. *P < 0.05, ***P < 0.001 versus sham
LXR agonist GW3965 induces target gene expression in cultured cardiomyocytes
Incubation of isolated adult cardiomyocytes with the synthetic LXR agonist, GW3965, for 20 h increased mRNA expression of the LXR target genes ABCA1, SREBP-1c and SCD1, by 10-, 2- and 2-fold, respectively (Fig. 3a). The responsiveness of the same target genes to GW3965 was investigated in HL-1 cells, and their mRNA expression nearly linearly increased with GW3965 concentrations ranging from 0.001 to 0.1 μM plotted on a logarithmic scale (Fig. 3b). The effect of GW3965 on target gene expression did not increase further with 1 μM (data not shown), but this concentration was used in succeeding experiments to facilitate activation of both LXRα and -β [14].

The synthetic LXR agonist GW3965 triggers target gene expression in cardiomyocytes. Levels of mRNA expression were determined by qRT-PCR and normalized to GAPDH in a adult cardiomyocytes isolated from mouse hearts (n = 3) incubated with 1 μM GW3965 or vehicle alone (control) for 18 h. *P < 0.05, ***P < 0.001 vs. Control; and b HL-1 cells cultured with different GW3965 concentrations for 18 h. Data are given as mean + SEM (n = 9). For SREBP-1c and ABCA1: ***P < 0.001; and for SCD1: ## P < 0.01, ### P < 0.001 versus vehicle control
LXR activation triggers lipid droplet formation in HL-1 cells
Incubation of HL-1 cells with GW3965 stimulated intracellular lipid accumulation both in the presence and absence of oleic acid, as evaluated by fluorescent lipid staining with Bodipy 493/503 (Fig. 4a). Quantitative analyses of the microscope images revealed that incubating HL-1 cells with GW3965 increased the number (Fig. 4b) and size distribution (see Supplemental Figure 1, Supplemental Material) of intracellular lipid droplets. In addition, analysis of cell lysates indicated that intracellular triglyceride levels were increased by GW3965 in the presence of oleic acid (Fig. 4c).

GW3965 causes enhanced lipid droplet formation in cardiomyocytes. HL-1 cells were incubated for 48 h in Claycomb medium in the presence of fatty acid (+FA; 500 μM oleic acid) or vehicle (−FA; 200 μM BSA alone), or in the presence of 1 μM LXR agonist (+GW3965) or vehicle (−GW39650; 0.1 % DMSO alone). a Cells were stained and representative confocal images (scale bar 100 μm) from the different conditions are shown: green lipid droplets (Bodipy 493/503); blue nuclei (Hoechst 33258). b The lipid droplets were identified and counted using ImageJ; for every experiment one image was taken of 3 replicate wells. Numbers represent cumulative values from 3 independently repeated experiments. c HL-1 cells were incubated using the same conditions as above, and cell lysates prepared for the measurements of total intracellular triglyceride (TG) levels. Bars represent mean + SEM (n = 9). *P < 0.05 versus negative control
Enhanced cardiac LXR target gene expression and lipid droplet formation in mice injected with GW3965
Mice were injected once with GW3965 or vehicle only to assess cardiac LXR activity in vivo. After 8 h, ABCA1, SREBP-1c and SCD1 mRNA levels increased 2- to 3-fold in left ventricles from mice injected with GW3965 compared to control mice (Fig. 5a). The mRNA levels of these genes were not significantly different 24 h after injection. At both time-points after GW3965 injections, mRNA levels of the LXRα and -β genes themselves were unchanged.

Liver X receptor (LXR) activation by the synthetic agonist GW3965 alters lipid metabolism in murine hearts. a C57BL/6 male mice were given a single i.p. injection of GW3965 or vehicle only. After 8 and 24 h, ventricular mRNA levels of SREBP-1c, ABCA1, SCD1 LXR target genes were determined by qRT-PCR and normalized to GAPDH mRNA levels. Bars represent group mean + SEM (n = 6–8). b Mice were injected i.p. with GW3965 or vehicle alone (control) and ventricles were sectioned and stained with Bodipy/Hoechst, and confocal images were obtained (scale bar 100 μm); green lipid droplets (Bodipy 493/503); blue nuclei (Hoechst 33258). Bars represent lipid droplet numbers quantified using the ImageJ software. c In addition, ventricular triglycerides (TG) were extracted, and their fatty acid profile determined by gas liquid chromatography. Bars represent the relative levels of different fatty acids, see also Supplementary figures; mean + SEM (n = 6). *P < 0.05, ***P < 0.001 versus negative control
The ability of GW3965 to affect intracellular lipid droplet accumulation in hearts was explored in vivo by staining cardiac cryosections with Bodipy 493/503 (Fig. 5b). Over the course of 24 h, multiple GW3965 injections were applied to sustain LXR activation. Quantification of microscope images showed that ventricles from these mice contained 76 % more lipid droplets as compared to control mice (Fig. 5b, lower panel).
To investigate if GW3965 also influenced triglyceride composition, fatty acid profiles in ventricular tissue was determined by gas liquid chromatography (Fig. 5c and Supplemental Table 1, Supplemental Material). In mice injected with GW3965, cardiac content of triglyceride 16:1Δ9 increased by 18 %, while 16:0 decreased by 13 %. In contrast, triglyceride 18:1Δ9, 18:0 and several other fatty acids were unchanged after GW3965 injections. These findings suggest that LXR activation is associated with a slight shift in cardiac triglyceride fatty acid composition in favor of monosaturates.
LXR activation alters cardiac expression of genes involved in triglyceride turnover and lipid droplet scaffolding
The effect of GW3965 on genes involved in triglyceride synthesis (ACSL3, GPAM, DGAT2), triglyceride degradation (ABDH5, PNPLA2) and lipid droplet scaffolding (PLIN2, PLIN5) was assessed in vivo and in vitro (Fig. 6). Mice injected with GW3965 displayed elevated expression of triglyceride synthesis genes ACSL3 and DGAT2, whereas triglyceride degradation genes ABDH5 (tendency; P = 0.08) and PNPLA2 were reduced. Lipid droplet-binding protein PLIN5 expression was reduced by 73 %, whereas PLIN2 expression was unchanged.

GW3965 alters cardiac expression of genes involved in triglyceride turnover and lipid droplet scaffolding. The upper panels show expression of genes involved in triglyceride synthesis, degradation, and lipid droplet formation in RNA extracts from left ventricles of C57BL/6 male mice 8 h after i.p. injection with GW3965 or vehicle only (mean + SEM; n = 6–8). The lower panels show expression of the same genes in RNA extracts from HL-1 cells incubated with 1 μM GW3965 for 18 h (mean + SEM; n = 4). Relative gene expression to GAPDH as endogenous control after qRT-PCR is shown for: acyl-CoA synthetase long-chain family member 3 (ACSL3); glycerol-3-phosphate acyltransferase (GPAM), mitochondrial; diacylglycerol O-acyltransferase 2 (DGAT2); abhydrolase domain containing 5 (ABHD5); patatin-like phospholipase domain containing 2 (PNPLA2); perilipin 2 (PLIN2); and perilipin 5 (PLIN5). *P < 0.05, **P < 0.01 versus control
Expression of the same set of genes was investigated in HL-1 cells and some of the findings in vivo were reproduced in this model. Cells incubated with GW3965 had increased expression of triglyceride synthesis genes ACSL3, GPAM and DGAT2 (tendency; P = 0.11). In contrast to the results in vivo, GW3965 had no effect on triglyceride degradation genes in HL-1 cells. The effect on lipid droplet-binding protein expression was different in the cells; PLIN2 increased, and PLIN5 expression was unchanged. The data suggest a link between lipid droplet formation by LXR in cardiac tissue and activation of the triglyceride synthesis pathway, and corresponding decline in the lipolytic pathways.
GW3965 modulates apoptotic response to hypoxia/reoxygenation in cardiomyocytes
Cardiomyocytes were incubated with GW3965 and subjected to hypoxia and reoxygenation. The proapoptotic markers Bax and activated Caspase 3 increased with hypoxia, whereas activated Caspase 3 alone was attenuated by GW3965 (Fig. 7a, c). Antiapoptotic marker Bcl-2 was unchanged with both hypoxia and GW3965 (Fig. 7a, c). The Bax:Bcl-2 ratio changed with hypoxia and tended to do so with GW3965 (P = 0.09), indicating that incubation with GW3965 may have an antiapoptotic influence on the cardiomyocytes (Fig. 7b). This finding gives support to a protective role of LXR activation after ischemia.

Modulation of the apoptosis pathways by GW3965 in HL-1 cells exposed to hypoxia/reoxygenation. HL-1 cells were preincubated with 1 μM GW3965 under normoxic conditions for 24 h prior to 2 h hypoxia followed by 8 h reoxygenation. Cells were harvested, proteins extracted and subjected to Western blotting using Ponceau as protein loading control. Antibodies against the proapoptotic Bax, the antiapoptotic Bcl2, and activated Caspase 3 were used. a Each bar represents quantification of Bax, Bcl-2 and Caspase 3 expression relative to ponceau (n = 3), and b calculation of the Bax:Bcl-2 apoptotic index. c Representative image of Western blots including Ponceau staining as loading control. Columns represent group mean ± SEM. **P < 0.01, difference due to hypoxia; #P < 0.05, difference due to GW3965
GW3965 injection in vivo attenuates the effects of ischemia–reperfusion in isolated hearts
Mice were injected with GW3965 or vehicle alone prior to heart isolation, Langendorff perfusion and functional assessment in a model of global ischemia and reperfusion. The post-ischemic increase of LVEDP observed in vehicle-treated hearts was attenuated by pretreatment of GW3965 (Fig. 8a). Furthermore, the post-ischemic depression of LVDP was attenuated in hearts of mice pretreated with GW3965. No statistical differences in HR or LVSP were observed between groups (data not shown). Coronary flow was slightly reduced during post-ischemic reperfusion, and this was not significantly influenced by GW3965 (Fig. 8b). When the infarct size was measured with TTC staining at the end of reperfusion, it was clearly reduced in hearts of mice pretreated with GW3965 (Fig. 8c). This indicates that ligand activation of LXR protects against ischemia–reperfusion injury in the isolated heart model.

LXR activation by GW3965 improves function and reduces infarct size in response to ischemia. Mice were injected i.p. with GW3965 (n = 7) or vehicle alone (control; n = 7); 6 h later, hearts were isolated, perfused, and 40 min global ischemia was induced followed by 60 min reperfusion. a Left ventricular end diastolic pressure (LVEDP), b left ventricular developed pressure (LVDP), c and coronary flow (CF) monitored before ischemia (BI) and at different time-points during reperfusion. d Infarct size evaluated in TCC stained heart slices at the end of reperfusion (infarcted area as % of total area). Data are presented as group mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus negative control
Discussion
The main findings of the present study were that LXRs are expressed in hearts, with an induced expression of LXRβ and target genes after in vivo infarction. Ligand stimulation with GW3965 dose-dependently increased LXR target gene expression in cardiac cells, and increased lipid droplet accumulation in cells and tissue. GW3965 also reduced ischemia–reperfusion injury in hearts isolated and perfused later.
LXRα and -β were both expressed in cardiac tissue, cardiomyocytes, cardiofibroblasts, and in HL-1 cells, with an abundance of LXRβ over LXRα. To our knowledge, this is the first study on LXR expression in the basal condition in cardiac cells. In a study of pressure overload through transaortic banding in wild type and LXRα knockout mice, sham-treated wild types had higher expression of LXRβ than LXRα, in accordance with the present findings [57]. The LXR agonist GW3965 affected the lipid composition in cardiomyocytes and heart, and induced expression of the LXR target genes ABCA1, SREBP-1c, and SCD1 in vitro and in vivo. The in vivo LXR-agonist activation was a rather rapid (<8 h) and transient effect. In addition, the same target genes were increasingly expressed in hearts 24 h post-infarction. This may suggest that LXR activity in the heart can be modulated both pharmacologically and physiologically. The target genes were chosen as markers of LXR activity on the basis of being bona fide target genes and representing different aspects of lipid metabolism. SREBP-1c is a transcription factor regulating genes involved in glucose utilization and fatty acid synthesis [9, 52]. ABCA1 is a transmembrane protein playing an important role in phospholipid homeostasis, regulating cholesterol and lipid removal from the cell [37]. SCD1 is a rate-limiting enzyme catalyzing the synthesis of monounsaturated fatty acids [17, 34]. Thus, the rapid and concurrent upregulation of these genes is likely to directly reflect LXR activation, and not secondary effects due to activation of other pathways.
Chronic overload of the fatty acid storage capacity in obesity may lead to aberrant fatty acid and hormonal release from visceral adipose tissues [18], paving the way to triglyceride accumulation in the heart and other non-adipose tissues. Cardiac triglyceride accumulation coincides under such conditions with several detrimental effects [5]. However, not only the amount, but also the type of fatty acids accumulating may be important, as well as the activation of nuclear receptors like the peroxisome proliferator-activated receptors or the LXRs. Here, we show that co-incubation with LXR agonist GW3965 and fatty acids induced accumulation of lipid droplets in HL-1 cells, and this was reflected in increased intracellular triglyceride levels. Similar results were found in cardiac tissue after in vivo GW3965 administration. Lipid droplets consist mainly of accumulated triglycerides, and these changes may be due to upregulated expression of the LXR target gene SREBP-1c, which induces lipogenic gene expression [50]. Interestingly, LXR ligand stimulation in vivo and in vitro increased expression of genes involved in triglyceride synthesis, indicating that LXR influences cardiac triglyceride synthesis. Genes regulating triglyceride degradation were downregulated after ligand stimulation in vivo, a finding which was not reproducible in HL-1 cells in vitro. In accordance with our results ACSL3 has been reported to be an LXR target gene [54]. The lipid droplet binding protein PLIN5 was downregulated after ligand stimulation in vivo, while PLIN2 was upregulated in vitro. PLIN2 has been reported to be a direct target of LXR [27]. The discrepancy between in vivo and in vitro findings after GW3965 stimulation can be interpreted as cell- and organ-specific effects of LXR-ligand stimulation. Furthermore, it indicates a role for LXR in regulation of lipid droplet scaffolding.
Detailed analysis of the Bodipy stained lipid droplets showed that HL-1 cells stimulated with GW3965 not only had larger lipid droplets, but that these also had a higher fluorescent intensity compared to controls, independent of the absence or presence of fatty acids in the media. We may speculate that the fluorescent properties or uptake into lipid droplets of the Bodipy stain are affected by the composition of lipid droplets. Cardiac SCD1 gene expression was enhanced by GW3965, which catalyzes the conversion of palmitic acid (16:0) and stearic acid (18:0) into mono-saturated palmitoleic acid (16:1Δ9) and oleic acid (18:1Δ9), respectively [17]. We predicted that the fatty acid profile would shift the ratio of unsaturated: saturated fatty acids. Indeed GW3965 increased relative levels of unsaturated fatty acid 16:1Δ9 to increase, and decreased saturated fatty acid 16:0 in vivo. This suggests that LXR-induced SCD1 activity, which affected lipid composition in the heart. Also in other tissues, upregulation of human SCD1 leads to a desaturation of saturated fatty acids [17, 34]. Saturated fatty acids may evoke ceramide accumulation and mitochondrial dysfunction [55]. SCD1 may also facilitate fatty acid esterification and storage, thereby preventing downstream effects of lipotoxicity [38]. It is possible that unsaturated fatty acids increases the incorporation of saturates in triglycerides [5]. Recently, it was reported that adenovirus-mediated SCD1 overexpression in neonatal rat cardiac myocytes induced lipid accumulation, also attenuating saturated fatty acid-induced apoptosis [30]. Cardiac lipids compartmentalized into droplets may have beneficial effects on cardiac function [5]. Thus, the concept that triglyceride accumulation in the heart is adverse to cardiac function may need modification.
The heart is a highly energy demanding organ in the body, normally generating 50–75 % of its ATP from oxidation of fatty acids. Easy accessible fatty acids, for instance from a high fat diet, may increase cardiac mitochondrial fatty acid oxidation [13]. Under circumstances such as myocardial ischemia, the heart switches the primary energy source to glucose instead of fatty acid, sparing the oxygen consumption in ATP generation [47]. Several studies indicate that excess free fatty acids are detrimental to cardiac function under ischemic conditions [51]. Lipid droplet accumulation as a result of fatty acid uptake is proposed to be a marker of ischemic injury in several organs [1]. However, recent evidence proposes that accumulation of lipid droplets can protect against apoptotic cell death by channeling free fatty acids into triglyceride pools [1, 32]. In support of this, we found that HL-1 cells subjected to hypoxia-reoxygenation had reduced expression of activated Caspase 3 and Bax after GW3965 stimulation. Furthermore, the lipid droplet associated protein PLIN5, when given to neonatal cardiomyocytes or rodent tissue, recruited mitochondria to the lipid droplet surface. This beneficially influenced lipid droplet hydrolysis and saturated fatty acid oxidation, protecting mitochondria [53]. Mice deficient of the gene encoding for PLIN5 had a decreased tolerance to oxidative stress and enhanced fatty acid oxidation, indicating a beneficial effect of lipid droplets in sequestering fatty acids from excessive oxidation [28]. The lipid droplet-binding protein PLIN5 was downregulated after GW3965 stimulation in vivo, however, a detailed study of the role of perilipins in myocardial infarction is warranted.
The final findings presented in this paper support that LXR activation may improve cardiac tolerance to ischemia. A variety of protein kinases are activated and interact at multiple levels and at various time-points during ischemia–reperfusion in the heart [22], which may lead to cell death. Activation of LXR blunted apoptotic response to hypoxia/reoxygenation in cardiomyocytes. Activated Caspase 3 increased with hypoxia, which was attenuated by GW3965. The proapoptotic marker Bax was also increased with hypoxia, but was unaffected by GW3965. Antiapoptotic marker Bcl-2 was unchanged with both hypoxia and GW3965. These data suggest that LXR activation has an antiapoptotic effect on cardiomyocytes in vitro. Furthermore, heart isolation and perfusion was performed after in vivo LXR agonism, at the time when lipid droplets had accumulated in cardiac tissue, and when LXR target genes as well as LXRβ itself were upregulated. Hearts of GW3965-stimulated mice subjected to global ischemia and reperfusion had improved heart function and reduced infarct size compared to hearts of vehicle-treated mice. It may be inferred from these experiments that GW3965-mediated gene transcription and lipid droplet accumulation in cardiac tissues prior to ischemia resulted in attenuation of injury. As ours is the first paper on LXR function in cardiac tolerance to ischemia, the discussion of our findings is limited to other, but related fields. One previous paper exists on the cardiac role of LXRα; in a model of pressure overload, LXRα null mice had an enhanced left ventricular hypertrophy [57]. Activation of the LXR heterodimerizing partner RXR by 9-cis RA increases glucose transporter GLUT4 expression in dedifferentiated rat cardiomyocytes [35], thus it is possible that LXR counteracts cardiac hypertrophy by enhancing glucose utilization. Ex vivo studies using a synthetic LXR ligand reduced cardiomyocyte hypertrophy and reduced activation of the transcription factor nuclear factor kappa B [57]. However, the pathophysiology underlying hypertrophy and acute myocardial infarction may be very different.
Limitations of the present study
The use of a synthetic LXR agonist does not reveal which isoform of LXR mediated the observed effects in this study. We did not do a detailed dose–response study of LXR agonist in vivo in relation to gene expression, lipid droplet accumulation, and heart function. Furthermore, in the present study we did not try to pinpoint all metabolic pathways involved when LXR leads to lipid droplet formation. Finally, the role of increased LXRβ and LXR target gene expression found in vivo myocardial infarction was not investigated further.
Conclusions
LXRs are expressed in the heart in the basal condition, and target gene expression is enhanced by myocardial ischemia. LXR agonist led to increased cardiac lipid droplet accumulation, changed the composition of triglycerides, and protected against induced ischemia- reperfusion.
References
Barba I, Chavarria L, Ruiz-Meana M, Mirabet M, Agullo E, Garcia-Dorado D (2009) Effect of intracellular lipid droplets on cytosolic Ca2+ and cell death during ischaemia–reperfusion injury in cardiomyocytes. J Physiol 587:1331–1341. doi:10.1113/jphysiol.2008.163311
Bilet L, van de Weijer T, Hesselink MK, Glatz JF, Lamb HJ, Wildberger J, Kooi ME, Schrauwen P, Schrauwen-Hinderling VB (2011) Exercise-induced modulation of cardiac lipid content in healthy lean young men. Basic Res Cardiol 106:307–315. doi:10.1007/s00395-010-0144-x
Boergesen M, Pedersen TA, Gross B, van Heeringen SJ, Hagenbeek D, Bindesboll C, Caron S, Lalloyer F, Steffensen KR, Nebb HI, Gustafsson JA, Stunnenberg HG, Staels B, Mandrup S (2012) Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor alpha in mouse liver reveals extensive sharing of binding sites. Mol Cell Biol 32:852–867. doi:10.1128/MCB.06175-11
Borradaile NM, Schaffer JE (2005) Lipotoxicity in the heart. Curr Hypertens Rep 7:412–417
Brindley DN, Kok BP, Kienesberger PC, Lehner R, Dyck JR (2010) Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol Endocrinol Metab 298:E897–E908. doi:10.1152/ajpendo.00509.2009
Broderick TL, Currie RW, Paulson DJ (1997) Heat stress induces rapid recovery of mechanical function of ischemic fatty acid perfused hearts by stimulating glucose oxidation during reperfusion. Can J Physiol Pharmacol 75:1273–1279
Calkin AC, Tontonoz P (2012) Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13:213–224. doi:10.1038/nrm3312
Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P (2001) A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 7:161–171. doi:10.1016/S1097-2765(01)00164-2
Chisholm JW, Hong J, Mills SA, Lawn RM (2003) The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44:2039–2048. doi:10.1194/jlr.M300135-JLR200
Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC (2012) Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 125:2844–2853. doi:10.1161/CIRCULATIONAHA.111.060889
Chu K, Miyazaki M, Man WC, Ntambi JM (2006) Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol 26:6786–6798. doi:10.1128/MCB.00077-06
Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA 95:2979–2984
Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N, Clarke K (2011) A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 106:447–457. doi:10.1007/s00395-011-0156-1
Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA, Willson TM (2002) Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 45:1963–1966
Crisafulli C, Di Paola R, Mazzon E, Paterniti I, Galuppo M, Genovese T, Bramanti P, Cappellani A, Cuzzocrea S (2010) Liver X receptor agonist treatment reduced splanchnic ischemia and reperfusion injury. J Leukoc Biol 87:309–321. doi:10.1189/jlb.0609438
Czibik G, Gravning J, Martinov V, Ishaq B, Knudsen E, Attramadal H, Valen G (2011) Gene therapy with hypoxia-inducible factor 1 alpha in skeletal muscle is cardioprotective in vivo. Life Sci 88:543–550. doi:10.1016/j.lfs.2011.01.006
Flowers MT, Ntambi JM (2008) Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr Opin Lipidol 19:248–256. doi:10.1097/MOL.0b013e3282f9b54d
Haugen F, Drevon CA (2007) The interplay between nutrients and the adipose tissue. Proc Nutr Soc 66:171–182. doi:10.1017/S0029665107005423
Haugen F, Zahid N, Dalen KT, Hollung K, Nebb HI, Drevon CA (2005) Resistin expression in 3T3-L1 adipocytes is reduced by arachidonic acid. J Lipid Res 46:143–153. doi:10.1194/jlr.M400348-JLR200
Heather LC, Clarke K (2011) Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol 50:598–605. doi:10.1016/j.yjmcc.2011.01.007
Hessvik NP, Boekschoten MV, Baltzersen MA, Kersten S, Xu X, Andersen H, Rustan AC, Thoresen GH (2010) LXRβ is the dominant LXR subtype in skeletal muscle regulating lipogenesis and cholesterol efflux. Am J Physiol Endocrinol Metab 298:E602–E613. doi:10.1152/ajpendo.00553.2009
Heusch G, Boengler K, Schulz R (2008) Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation 118:1915–1919. doi:10.1161/CIRCULATIONAHA.108.805242
Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 383:728–731. doi:10.1038/383728a0
Jodalen H, Stangeland L, Grong K, Vik-Mo H, Lekven J (1985) Lipid accumulation in the myocardium during acute regional ischaemia in cats. J Mol Cell Cardiol 17:973–980 pii:S0022-2828(85)80077-8
Juvet LK, Andresen SM, Schuster GU, Dalen KT, Tobin KA, Hollung K, Haugen F, Jacinto S, Ulven SM, Bamberg K, Gustafsson JA, Nebb HI (2003) On the role of liver X receptors in lipid accumulation in adipocytes. Mol Endocrinol 17:172–182
King LM, Opie LH (1998) Glucose and glycogen utilisation in myocardial ischemia–changes in metabolism and consequences for the myocyte. Mol Cell Biochem 180:3–26
Kotokorpi P, Venteclef N, Ellis E, Gustafsson JA, Mode A (2010) The human ADFP gene is a direct liver-X-receptor (LXR) target gene and differentially regulated by synthetic LXR ligands. Mol Pharmacol 77:79–86. doi:10.1124/mol.109.059063
Kuramoto K, Okamura T, Yamaguchi T, Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K, Usuda N, Matsumura S, Inoue K, Fushiki T, Kojima Y, Hashimoto T, Sakai F, Hirose F, Osumi T (2012) Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J Biol Chem 287:23852–23863. doi:10.1074/jbc.M111.328708
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258. doi:10.1152/physrev.00015.2009
Matsui H, Yokoyama T, Sekiguchi K, Iijima D, Sunaga H, Maniwa M, Ueno M, Iso T, Arai M, Kurabayashi M (2012) Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS ONE 7:e33283. doi:10.1371/journal.pone.0033283
McGavock JM, Victor RG, Unger RH, Szczepaniak LS, American College of P, the American Physiological S, (2006) Adiposity of the heart, revisited. Ann Intern Med 144:517–524. pii:144/7/517
Menendez JA, Colomer R, Lupu R (2004) Inhibition of tumor-associated fatty acid synthase activity enhances vinorelbine (Navelbine)-induced cytotoxicity and apoptotic cell death in human breast cancer cells. Oncol Rep 12:411–422
Mitro N, Vargas L, Romeo R, Koder A, Saez E (2007) T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett 581:1721–1726. doi:10.1016/j.febslet.2007.03.047
Miyazaki M, Ntambi JM (2003) Role of stearoyl-coenzyme A desaturase in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids 68:113–121
Montessuit C, Papageorgiou I, Campos L, Lerch R (2006) Retinoic acids increase expression of GLUT4 in dedifferentiated and hypertrophied cardiac myocytes. Basic Res Cardiol 101:27–35. doi:10.1007/s00395-005-0567-y
O’Connell TD, Rodrigo MC, Simpson PC (2007) Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol 357:271–296. doi:10.1385/1-59745-214-9:271
Ouvrier A, Cadet R, Vernet P, Laillet B, Chardigny JM, Lobaccaro JM, Drevet JR, Saez F (2009) LXR and ABCA1 control cholesterol homeostasis in the proximal mouse epididymis in a cell-specific manner. J Lipid Res 50:1766–1775. doi:10.1194/jlr.M800657-JLR200
Peter A, Weigert C, Staiger H, Rittig K, Cegan A, Lutz P, Machicao F, Haring HU, Schleicher E (2008) Induction of stearoyl-CoA desaturase protects human arterial endothelial cells against lipotoxicity. Am J Physiol Endocrinol Metab 295:E339–E349. doi:10.1152/ajpendo.00022.2008
Phelan CA, Weaver JM, Steger DJ, Joshi S, Maslany JT, Collins JL, Zuercher WJ, Willson TM, Walker M, Jaye M, Lazar MA (2008) Selective partial agonism of liver X receptor alpha is related to differential corepressor recruitment. Mol Endocrinol 22:2241–2249. doi:10.1210/me.2008-0041
Reue K (2011) A thematic review series: lipid droplet storage and metabolism: from yeast to man. J Lipid Res 52:1865–1868. doi:10.1194/jlr.E020602
Ruusalepp A, Czibik G, Flatebo T, Vaage J, Valen G (2007) Myocardial protection evoked by hyperoxic exposure involves signaling through nitric oxide and mitogen activated protein kinases. Basic Res Cardiol 102:318–326. doi:10.1007/s00395-007-0644-5
Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B (2000) Role of LXRs in control of lipogenesis. Genes Dev 14:2831–2838. doi:10.1101/Gad.850400
Sironi L, Mitro N, Cimino M, Gelosa P, Guerrini U, Tremoli E, Saez E (2008) Treatment with LXR agonists after focal cerebral ischemia prevents brain damage. FEBS Lett 582:3396–3400. doi:10.1016/j.febslet.2008.08.035
Skyschally A, Schulz R, Heusch G (2008) Pathophysiology of myocardial infarction: protection by ischemic pre- and postconditioning. Herz 33:88–100. doi:10.1007/s00059-008-3101-9
Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ (2010) PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation. J Clin Invest 120:3443–3454. doi:10.1172/JCI40905
Sparrow CP, Baffic J, Lam MH, Lund EG, Adams AD, Fu X, Hayes N, Jones AB, Macnaul KL, Ondeyka J, Singh S, Wang J, Zhou G, Moller DE, Wright SD, Menke JG (2002) A potent synthetic LXR agonist is more effective than cholesterol loading at inducing ABCA1 mRNA and stimulating cholesterol efflux. J Biol Chem 277:10021–10027. doi:10.1074/jbc.M108225200
Stanley WC, Lopaschuk GD, Hall JL, McCormack JG (1997) Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 33:243–257
Straeter-Knowlen IM, Evanochko WT, den Hollander JA, Wolkowicz PE, Balschi JA, Caulfield JB, Ku DD, Pohost GM (1996) 1H NMR spectroscopic imaging of myocardial triglycerides in excised dog hearts subjected to 24 hours of coronary occlusion. Circulation 93:1464–1470
Suzuki J, Shen WJ, Nelson BD, Patel S, Veerkamp JH, Selwood SP, Murphy GM Jr, Reaven E, Kraemer FB (2001) Absence of cardiac lipid accumulation in transgenic mice with heart-specific HSL overexpression. Am J Physiol Endocrinol Metab 281:E857–E866
Tontonoz P, Kim JB, Graves RA, Spiegelman BM (1993) ADD1: a novel helix–loop–helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol 13:4753–4759
Vary TC, Reibel DK, Neely JR (1981) Control of energy metabolism of heart muscle. Annu Rev Physiol 43:419–430. doi:10.1146/annurev.ph.43.030181.002223
Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK (2003) Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol 23:5780–5789
Wang H, Sreenevasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52:2159–2168. doi:10.1194/jlr.M017939
Weedon-Fekjaer MS, Dalen KT, Solaas K, Staff AC, Duttaroy AK, Nebb HI (2010) Activation of LXR increases acyl-CoA synthetase activity through direct regulation of ACSL3 in human placental trophoblast cells. J Lipid Res 51:1886–1896. doi:10.1194/jlr.M004978
Wende AR, Abel ED (2010) Lipotoxicity in the heart. Biochim Biophys Acta 1801:311–319. doi:10.1016/j.bbalip.2009.09.023
Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, Miller AB, Moore J, McKee DD, Moore L, Nichols J, Parks D, Watson M, Wisely B, Willson TM (2003) X-ray crystal structure of the liver X receptor beta ligand binding domain: regulation by a histidine–tryptophan switch. J Biol Chem 278:27138–27143. doi:10.1074/jbc.M302260200
Wu S, Yin R, Ernest R, Li Y, Zhelyabovska O, Luo J, Yang Y, Yang Q (2009) Liver X receptors are negative regulators of cardiac hypertrophy via suppressing NF-κB signalling. Cardiovasc Res 84:119–126. doi:10.1093/cvr/cvp180
Acknowledgments
This work was financially supported by The Norwegian Women’s Public Health Association (Norske Kvinners Sanitetsforening), The Norwegian Health Association (Nasjonalforeningen for folkehelsen), Novo Nordisk Foundation, UNIFOR (Legatet til Henrik Homans Minde, S.G. Sønneland Foundation, Anders Jahres fond til vitenskapens fremme, Aktieselskabet Freia Chocolade Fabriks Medisinske Fond) and The University of Oslo. Peng Lei was supported by the China Scholarship Council.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lei, P., Baysa, A., Nebb, H.I. et al. Activation of Liver X receptors in the heart leads to accumulation of intracellular lipids and attenuation of ischemia–reperfusion injury. Basic Res Cardiol 108, 323 (2013). https://doi.org/10.1007/s00395-012-0323-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0323-z