
Abstract
Postconditioning (Postcon) reduces infarct size. However, its role in modulation of cardiac repair after infarction is uncertain. This study tested the hypothesis that Postcon inhibits adverse cardiac repair by reducing degradation of extracellular matrix (ECM) and synthesis of collagens via modulating matrix metalloproteinase (MMP) activity and transforming growth factor (TGF) β1/Smad signaling pathway. Sprague–Dawley rats were subjected to 45 min ischemia followed by 3 h, 7 or 42 days of reperfusion, respectively. In acute studies, four cycles of 10/10 s Postcon significantly reduced infarct size, which was blocked by administration of a mitochondrial KATP channel blocker, 5-hydroxydecanoate (5-HD) at reperfusion. In chronic studies, Postcon inhibited MMP activity and preserved ECM from degradation as evidenced by reduced extent of collagen-rich scar and increased mass of viable myocardium. Along with a reduction in collagen synthesis and fibrosis, Postcon significantly down-regulated expression of TGFβ1 and phospho-Smad2/3, and up-regulated Smad7 as compared to the control, consistent with a reduction in the population of α-smooth muscle actin expressing myofibroblasts within the infarcted myocardium. At 42 days of reperfusion, echocardiography showed significant improvements in left ventricular end-diastolic volume and ejection fraction. The wall thickness of the infarcted middle anterior septum in the Postcon was also significantly greater than that in the control. The beneficial effects of Postcon on cardiac repair were comparable to preconditioning and still evident after a blockade with 5-HD. These data suggest that Postcon is effective to promote cardiac repair and preserve cardiac function; protection is potentially mediated by inhibiting ECM degradation and collagen synthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite the considerable progress in treatment and management of ischemic heart disease in last three decades, acute myocardial infarction is still the leading cause of patient’s mortality after coronary occlusion [29]. Myocardial salvage by timely restoration of blood flow (i.e., reperfusion) to the infarct-related artery is associated with smaller infarct size, less enzyme release and better cardiac function recovery in patients after ischemia [47, 57]. Given the fact that reperfusion also elicits a broad range of injury pathologies paradoxically leading to variable amounts of salvageable myocardium [1, 54], the means and timing of restoring optimal blood perfusion continue to be a highly debated and studied topic [21, 38, 44].
Postconditioning (Postcon), the rapid sequential intermittent interruptions of blood flow applied during early moments of reperfusion has been shown to attenuate myocardial injury in a variety of animal models since we formerly reported its sparing effect on infarct size in 2003 [59]. Postcon is known to alter the formation of reactive oxygen species [17, 34], stimulate survival kinases such as p42/44 ERK and PI-3K-Akt [16, 53], slow down recovery of tissue pH [8], activate mitochondrial KATP channels [32, 36, 37] and inhibit mitochondrial permeability transition pore opening by activating STAT3 pathway [4, 20]. Consistent with the findings from animal studies, clinical observations have also revealed the inhibitory effects of Postcon on magnitude of ST-segment elevation, release of myocardial enzyme and induction of infarction in patients undergoing percutaneous coronary intervention [47, 52, 57] or coronary artery bypass graft surgery [30, 43].
Adverse cardiac repair collateral to myocardial infarction occurs during extended phases of reperfusion, characterized by progressive infarct expansion, ventricular wall thinning and chamber dilation [35, 60]. These processes encompass degradation of native ECM and healing of the infarct during which fibroblasts proliferate and deposit collagen to form a reparative fibrosis and a non-contractile scar, resulting in further ventricular dilation, cardiac dysfunction and heart failure [2, 3, 5, 28, 31, 46].
Clinical studies have previously yielded encouraging data showing that protection with Postcon on infarct size, myocardial blood flow and cardiac function is sustained, and still detected when patients were re-examined at several months or years after treatment, suggesting that Postcon can afford persistent benefits on cardiac repair after myocardial infarction [14, 25, 26, 52]. However, we do not know whether these beneficial effects of Postcon against myocardial injury are associated with the stimulation of an endogenous repair process responsible for a favorable cardiac repair. Therefore, we tested the hypothesis that Postcon inhibits fibrotic process and improves cardiac function in the rat model of ischemia/reperfusion-induced heart failure. Specifically, the effects of Postcon on inflammatory response, MMP activity, ECM degradation and TGFβ1/Smad-mediated collagen synthesis were examined. To determine whether the promotion of cardiac repair with Postcon is independent of infarct size reduction, a putative KATP channel blocker, 5-HD that we have previously shown its blocking effect on infarct size by Postcon [32], was administered before Postcon. Furthermore, to demonstrate the efficacy of Postcon on cardiac repair, this study was also compared with conventional pre-conditioning (Precon).
Materials and methods
Surgical preparation of animals
All animals received humane care in compliance with ‘The Guide for the Care of Use of Laboratory Animals” published by the US National Institute of Health (NIH Publication No. 85–23, revised 1996). Male Sprague–Dawley rats weighing 400–450 g were anesthetized with an intraperitoneal injection of a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg). The animals were intubated and mechanically ventilated with oxygen-enriched room air using a rodent respirator. The chest was opened by a left thoracotomy through the fourth intercostal space. After pericardiotomy, a 6-0 polyproline ligature was placed under the left coronary artery (LCA), where it emerged from beneath the left atrium, and the ends of the tie were threaded through a small plastic tube to form a snare for reversible LCA occlusion. At the end of the surgical operation, the incisions were closed in layers. The chest and endotracheal tubes were removed after the spontaneous breathing was recovered. Postoperative analgesia was done by injecting buprenorphine (0.1 mg/kg, i.p) for 3 days.
Experimental protocol
The whole study was divided into two protocols (Fig. 1). In protocol I, four groups (n = 7/group) were randomly assigned to demonstrate the effects of Postcon on infarct size and cytokine production. In the control group, the rats were subjected to 45 min ischemia followed by 3 h of reperfusion; in the Postcon group, four cycles of 10/10 s reperfusion/ischemia were applied at the onset of reperfusion; in the 5-HD + Postcon group, 5-HD (Sigma Chemicals, St. Louis, MO, USA) was injected intravenously at a dose of 10 mg/kg 5 min before Postcon [32]; in the 5-HD group, 5-HD was injected intravenously only at reperfusion. In protocol II, the rats in five groups (n = 8/group at each time point) were subjected to 45 min ischemia followed by 7 or 42 days of reperfusion, respectively; in the Postcon and Precon groups, four cycles of 10/10 s reperfusion/ischemia and two cycles of 5/5 min ischemia/reperfusion were selected as algorithms of Postcon and Precon, respectively; In the 5-HD + Postcon group, 5-HD was injected intravenously 5 min before Postcon; In the Sham group (n = 4 at each time point), the chest was opened and no LCA occlusion was conducted during the experiment. In all rats, heparinization was conducted with a bolus injection of 200 U/kg sodium heparin prior to LCA occlusion.

Study was divided into two parts: in protocol I (n = 7/each group): in control, rats were subjected to 45 min ischemia and 3 h of reperfusion; in Postcon, four cycles of 10/10 s reperfusion/ischemia were applied at reperfusion; in 5-HD + Postcon, a mitochondrial KATP channel blocker, 5-HD was infused at 5 min before Postcon; in 5-HD, drug was given alone. In protocol II (n = 8/each time point): in control, rats were subjected 45 ischemia followed by 7 or 42 days of reperfusion, respectively; in Postcon and 5-HD + Postcon, algorithm of Postcon and 5-HD dose were same as those in Protocol I; in Precon, two cycles of 5/5 min ischemia/reperfusion were applied before ischemia; in Sham (n = 4/each time point), the chest was opened without ischemia. At the end of the experiment, the heart was removed for histological analysis after cardiac function was measured by echocardiograph
Determination of area at risk and infarct size
At the end of 3 h of reperfusion in studies of protocol I, the LCA was re-ligated and Unisperse blue dye was injected into the carotid vein to stain the non-ischemic region blue and thereby outline the area at risk (Ar). The Ar was separated from the non-ischemic zone and incubated in a 1 % solution of triphenyltetrazolium chloride at 37 °C to differentiate the area of necrosis (An) from the non-necrotic Ar. The Ar, as a percent of the left ventricular mass (Ar/LV), and the An, as a percent of the Ar (An/Ar, infarct size) were calculated by tissue weight as we reported previously [26, 59].
Detection of plasma TNFα and IL-6
Immunoreactive TNFα and IL-6 levels were determined with an Elisa kit (R and D Systems, Minneapolis, MN, USA) according to the manufacturer’s instruction. In brief, the plasma extracted from the carotid vein was reacted with the assay reagents in TNFα and IL-6 kits, respectively, and analyzed spectrophotometrically (BioTek Epoch Microplate Spectrophotometer, Winooski, VT) at 450 nm of absorbance. Levels of TNFα and IL-6 were calculated from the standards and expressed as μg/ml of plasma [26].
Detection of collagen deposition by Masson’s trichrome staining
Masson’s trichrome staining was used to evaluate collagen deposition within the scar zone as we previously reported [62]. In brief, the paraffin sections were deparaffinized, hydrated with distilled water and stained with Masson’s trichrome method. The staining produces collagen blue, nuclei black and viable muscle fiber red. Eight randomized high-powered fields per tissue section were averaged to determine the collagen-rich area calculated as the percentage of the entire left ventricular (LV) scar zone. Images were processed using a digital image analyzer (ImageJ, NIH, Bethesda, MA, USA).
Determination of MMP activity by gelatin zymography
Gelatin zymography was performed as described previously [12]. In brief, freshly frozen tissue samples were homogenized in lysis buffer and the gelatin (Sigma Chemical Co., St. Louis, MO) was used as a substrate. Samples (20 μg) and zymograghy standard (7 ng) of purified human MMP-9 and MMP-2 (Chemicon Inc. Temecula, CA, USA) were directly loaded on to standard polyacrylamide gels. After incubation, the gels were stained in Coomassie blue R-250 and distained in methanol and acetic acid until clear bands were displayed. Molecular size of each band displaying enzymatic activity was characterized by comparison with purified standard MMP-9 or MMP-2. Zymographic activity was quantified using a digital image analyzer (ImageJ, NIH, Bethesda, MA).
Expression of TGFβ1, collagen and Smads by Western blotting
Western blotting was performed as we described previously [62]. In brief, the freshly frozen tissue samples were homogenized in lysis buffer and protein concentration was measured by the DC protein assay. The protein was then boiled and loaded on to gradient SDS-polyacrylamide gel using Mini Protean II Dual Stab Cell (Bio Rad). Membranes were subsequently exposed to the following antibodies: the mouse monoclonal anti-TGFβ1 (Abcam Inc. Cambrige, MA, USA), the goat polyclonal anti-collagen I and the mouse monoclonal anti-collagen III (Sigma, St. Louis, MO), the rabbit monoclonal phospho-Smad2 and Smad3 (Cell Signaling, Danvers, MA, USA), the rabbit polyclonal anti-Smad4 and Smad7 (Santa Cruz Biotech, CA, USA), respectively. Bound antibody was detected by horseradish peroxidase conjugated anti-rabbit IgG. The membrane was incubated with chemiluminescence substrate and exposed to an X-ray film. The scanned images were imported into the ImageJ. Actin was used as a standard of protein-loading control for normalizing bands at different time points. The final results are calculated as the ratio of intensity of each band divided by actin intensity.
Differentiation of fibroblasts with immunohistochemistry
Immunohistochemical staining on tissue sections was performed as we described previously [62]. In brief, paraffin-embedded blocks were deparaffinized in xylene and dehydrated in graded ethanol. The transverse paraffin sections were stained using a monoclonal antibody against α-smooth muscle actin (SMA, Sigma Chemical Co., St. Louis, MO). Quality of the assay was controlled by either elimination of the primary antibody or incubation of the tissue with a non-immune IgG. Data were analyzed using computed-assisted morphometry (ImageJ, NIH, Bethesda, MA). Differentiation of fibroblasts was reported as a mean number of α-SMA—expressing myofibroblasts from eight randomized high-powered fields.
Cardiac function and wall thickness by echocardiography
Echocardiography was used to assess LV systolic and diastolic function using two-dimensional (2D) guided M-mode ultrasound system (Acuson Sequoia, Siemens Medical Solutions Inc. CA, USA) via a 15-MHz linear transducer as we previously reported [62]. In brief, LV systolic and diastolic function were measured by calculating fraction shortening (FS), ejection fraction (EF) and rapid early LV filling, E-wave velocity/atrial contraction filling, A-wave velocity (E/A ratio), respectively. 2D images, which were frozen at the end of diastole were used to measure LV volume and interventricular septum/posterior wall thickness. The echocardiography was performed before opening the chest (baseline) and during the time course of the experiment in all groups. All measurements were averaged over three consecutive cardiac cycles.
Statistical analysis
All data were reported as mean ± standard error. An one-way ANOVA followed by Student–Newman–Keul’s post hoc test was used to analyze group differences in the level of cytokines, intensity of TGFβ1, collagens, Smads, percentage of collagen-rich area and population of fibroblast differentiation. Echocardiographic data were analyzed by one-way repeated measures ANOVA followed by post hoc analysis with Student–Newman–Keul’s test for multiple comparisons by SigmaStat (Systat Software Inc., Point Richmond, CA, USA). Statistical significance was set at a value of p < 0.05.
Results
Changes in plasma TNFα and IL-6 during reperfusion
Changes in plasma TNFα and IL-6 levels are shown in Fig. 2a. Relative to the ischemia, reperfusion caused a significant elevation in TNFα with a peak at 1 h and a constant increase in IL-6 during the observational period in the control group. However, these changes in levels of TNFα and IL-6 were significantly inhibited by Postcon. 5-HD administered 5 min before reperfusion completely blocked the inhibitory effect of Postcon on TNFα and IL-6 levels, but these changes were not altered by 5-HD alone.

Measurement of cytokines and infarct size. a Levels of plasma TNFα and IL-6 during the course of the experiment. Postcon (Post) significantly reduced both TNFα and IL-6 levels during reperfusion, but inhibition was not blocked by 5-HD. b Area at risk (AAR), expressed as a percentage of the left ventricle and area of necrosis (infarct size), expressed as a percentage of the AAR. Post and Precon (Pre) significantly reduced infarct size at the end of 3 h of reperfusion, but its sparing effect on infarct size was blocked by 5-HD. Values are mean ± SEM. n = 7/group. * p< 0.05 versus baselines and ischemia; † p < 0.05 versus control
Area at risk and infarct size
No difference in the area at risk myocardium among all groups was found (Fig. 2b). Infarct size in the Postcon and Precon groups was 19 ± 3 and 21 ± 4 % less than that in the control group (34 ± 2, 33 ± 3 vs. 42 ± 6 %, p < 0.05), respectively. Infusion of 5-HD before reperfusion did not change the extent of the area at risk, but it completely blocked the sparing effect of Postcon on infarct size. 5-HD administered alone had no effect on infarct size relative to the control.
Collagen deposition in the scar tissue
The area of collagen deposition in the infarcted scar tissue was evaluated using Masson’s trichrome staining. As shown in Fig. 3, no newly synthesized collagen was detected in the non-infarct zone among all groups. However, the collagen-rich area through the entire ventricular wall from endomyocardium to epimyocardium was extended over reperfusion, and less viable myocardium was detected at day 42 in the control animals. The hearts treated with Postcon and Precon exhibited a significant reduction in collagen deposition within the infarcted scar zone, as evidenced by smaller collagen-rich area when compared with the control group. The ischemic/reperfused myocardium appeared more organized and circumscribed in these two groups, suggesting a favorable cardiac repair. Infusion of 5-HD prior to reperfusion did not reverse the inhibitory effect of Postcon on collagen deposition during 42 days of observation. No collagen deposition was detected in the Sham group (data not shown).

Identification of collagen deposition and fibrotic tissue formation by Masson’s trichrome staining. There was extensive loss of myocardial mass at 42 days of reperfusion in control. However, formation of collagen deposition and fibrotic tissue was significantly reduced with increased mass of viable myocardium in Postcon and Precon, respectively. Infusion of 5-HD did not alter beneficial effect of Postcon on tissue repair. The collagen-rich area calculated as the percentage of the area at risk myocardium was shown on the right panel. Endo endomyocardium, Epi epimyocardium. Values are mean ± SEM. n = 8/group. * p < 0.05 versus 7-day; † p < 0.05 versus control. Original magnifications: 100×
Change in activity of MMPs
Activity of MMPs on gelatin zymography is shown in Fig. 4. pro-MMP-9, but not MMP-9, was clearly expressed in the control group. Consistent with the change in expression of pro-MMP-2, activity of MMP-2 was markedly increased relative to the non-ischemic zone in the control group, suggesting activation of pro-form during reperfusion. However, pro-MMP-9, pro-MMP-2 and MMP-2 were significantly inhibited by Postcon and Precon, demonstrating effective inactivation of MMPs with these interventions. 5-HD administered 5 min prior to reperfusion had no effect on Postcon attenuated activity of MMPs. There was no significant difference in expression of MMPs during the course of the experiment between the non-ischemic myocardium in the control and those in the Sham group (data not shown).

Activity of MMPs by gelatin zymography. MMP-9 was expressed at the 92-kDa band (pro-form) and the 84-kDa (active form); MMP-2 was evident at the 72-kDa (pro-form) and the 62-kDa (active form) during reperfusion. Postcon and Precon significantly reduced the activity of both pro- and active forms of MMP-2 during reperfusion, but inhibition by Postcon was not altered by 5-HD as measured by arbitrary unit on the right panel. Values are mean ± SEM. n = 8/group. * p < 0.05 versus normal and non-ischemic zone; † p < 0.05 versus control
Changes in collagen synthesis
To further confirm the results demonstrated by Masson’s trichrome staining, we measured the expression of collagen types I and III in the transmural tissue samples of the area at risk myocardium using Western blotting as shown in Fig. 5. Collagen types I and III were expressed in the non-ischemic zone in the control group and normal tissue in the Sham group (data not shown), but no statistical difference between groups was detected. However, the synthesis of these collagens was significantly increased at day 7, and continuously maintained at higher levels at day 42 relative to the non-ischemic zone in the control group. The hearts treated with Postcon and Precon comparatively reduced collagen levels relative to the control group at all-time points measured. 5-HD administration before reperfusion did not alter the inhibitory effect of Postcon on collagens. These data were consistent with findings showing a reduction in fibrotic tissue in the infarcted myocardium identified by Masson’s trichrome staining.

Expression of collagen I, III and Smad2 by Western blot. Relative to the non-infarct tissue, significant increase in levels of collagens I, III, Smad2 and phosphor-Smad2 was detected during reperfusion in control. However, these changes were significantly attenuated by Postcon and Precon over time of the observation. The protection was not altered by 5-HD. All bands were normalized by actin as shown at the bottom of each panel. Values are mean ± SEM. n = 8/group. * p < 0.05 versus non-ischemia zone (N); † p < 0.05 versus control
Changes in phosphorylation of Smads and expression of TGFβ1
To demonstrate whether inhibition of fibrotic tissue formation by Postcon is associated with TGFβ1/Smads signaling pathway, we measured Smad phosphorylation and TGFβ1 expression from the transmural tissue samples obtained from the area at risk myocardium using Western blotting assay. As shown in Figs. 5 and 6, total protein of Smad2, Smad4 and Smad7, but not Smad3 was detected in the non-ischemic zone, however, no statistical difference was found among all groups. Ischemia/reperfusion caused a significant increase in expression of total Smad2 and Smad3 as well as their phosphorylated forms during the course of the observation, consistent with enhanced expression of Smad4. Furthermore, expression of Smad7 was down-regulated during the course of the experiment. Postcon and Precon comparatively abrogated phosphorylation of Smad2 and Smad3 as well as expression of Smad4, and up-regulated Smad7. As shown in Fig. 7a, TGFβ1 was expressed in the non-ischemic zone in the control group and normal tissue in the Sham group (data not shown), but no statistical difference between groups was found. Ischemia/reperfusion caused a constant increase in TGFβ1 expression during the course of the experiment, but, this change was significantly inhibited by Postcon and Precon. 5-HD administration did not change inhibition of Postcon on expression of Smads and TGFβ1. These results were consistent with significant reduction of collagen synthesis/deposition in the infarcted myocardium detected by Western blotting assay and Masson’s trichrome staining.

Expression of Smads by Western blot. Relative to the non-infarct tissue, ischemia/reperfusion up-regulated Smad3, phosphor-Smad3, Smad4, but down-regulated Smad7 in control. However, these changes were significantly modulated by Postcon and Precon over time of the observation. The expression of Smads was not blocked by 5-HD. All bands were normalized by actin as shown at the bottom of each panel. Values are mean ± SEM. n = 8/group. * p < 0.05 versus non-ischemia zone (N); † p < 0.05 versus control

Expression of TGFβ1 and accumulation of α-SMA expressing myofibroblasts. Ischemia/reperfusion up-regulated TGFβ1 expression (a) and increased the number of α-SMA expressing myofibroblasts (b) over time of reperfusion in control. However, these changes were significantly inhibited by Postcon and Precon, but not blocked by 5-HD. Values are mean ± SEM. n = 8/group. * p < 0.05 versus non-ischemia zone (N) and 7 day; † p < 0.05 versus control
Differentiation of fibroblast in myocardium
To understand potential mechanism involved in induction of collagens, we detected the accumulation of α-SMA—expressing myofibroblasts, a marker of fibroblast differentiation, using immunohistochemistry. As shown in Fig. 7b, a few myofibroblasts were present only in vascular smooth muscle cells in the non-ischemic myocardium among all experimental groups. However, myofibroblasts were significantly increased after 7 days of reperfusion in the infarcted zone in the control group. The majority of myofibroblasts had aligned with the host myocardial fibers and along the ischemic border and scar zones. At 42 days of reperfusion, the number of myofibroblasts was still significantly higher in the control group relative to the non-ischemic zone and Sham control (data not shown). However, accumulation of myofibroblasts in the infarcted zone in the Postcon and Precon group was significantly reduced during the entire period of reperfusion, suggesting an inhibition of fibroblast differentiation. 5-HD administration did not alter inhibitory effect of Postcon on fibroblast differentiation.
Evaluation of cardiac systolic and diastolic function
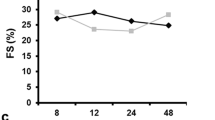
Echocardiographic results of cardiac systolic and diastolic function among groups are summarized in Table 1. No significant statistical difference was found in all parameters measured in the Sham and baseline values among groups, so data from these groups were averaged. During 42 days of reperfusion in the control group, LVDd, LVDs, EDV and ESV were significantly greater than baseline values, but, the indexes of systolic function, i.e., FS and SV were significantly reduced, suggesting ventricular contractile dysfunction (Fig. 8). However, these parameters were improved in the hearts treated with Postcon and Precon over the course of the experiment. EF values increased by 24 ± 4 % in Postcon and 29 ± 3 % in Precon, respectively, as compared to the control group at 42 days of reperfusion. An example of pulsed-wave Doppler recordings of mitral inflow is shown in Fig. 9a. Ischemia/reperfusion resulted in a significant decrease in the E/A ratio relative to baseline values, largely due to an increased A wave during reperfusion in the control group, suggesting an impaired relaxation. However, this change in E/A ratio was inhibited by Postcon and Precon. 5-HD administration before reperfusion did not reverse the protective effects of Postcon on cardiac systolic and diastolic function.

LV systolic function. Ischemia/reperfusion caused significant reduction in cardiac systolic function as measured from a short axis view of LV at the level of the papillary muscles over 42 days of reperfusion. Relative to these values in control, Postcon and Precon significantly preserved cardiac systolic function, but protection with Postcon was not altered by 5-HD. FS fraction shortening, EF ejection fraction. Values are mean ± SEM. n = 8/group. * p < 0.05 versus respective baselines among all groups; † p < 0.05 versus control

LV diastolic function and wall thickness. Cardiac diastolic function was measured by a ratio of E-wave velocity and A-wave velocity (a). The wall thickness was measured from a short view of LV at the level of the papillary muscles (b). Relative to control, Postcon and Precon significantly preserved E/A ratio (>2 E/A ratio >1 in normal), reduced end-diastolic volume and increased wall thickness in infarcted middle anterior and septal myocardium during reperfusion. The protection was not altered by 5-HD. Values are mean ± SEM. n = 8/group. * p < 0.05 versus baseline values in control; † p < 0.05 versus control
Cardiac repair
The LV end-diastolic dimension and wall thickness were used to assess cardiac repair with Postcon. As shown in Fig. 9b, LV end-diastolic volume at baseline was comparable during the course of the experiment among groups. However, ischemia/reperfusion significantly increased LV end-diastolic dimension when compared with the baseline in the control groups. Furthermore, the wall thickness of the infarct middle anterior septum was significantly reduced relative to the baseline during reperfusion in the control group. Consistent with an improvement of cardiac function in the Postcon group, the hearts treated with Postcon had smaller LV dimension, as assessed by LV end-diastolic volume, and greater wall thickness of the infarcted middle anterior septum, as measured from echocardiographic images, relative to that in the control group (1.6 ± 0.03 mm in Postcon, 1.5 ± 0.02 mm in Precon, 1.6 ± 0.04 mm in 5-HD + Post vs. 1.3 ± 0.01 mm in control, respectively, all p < 0.01).
Discussion
The results of the present study demonstrated a promotion of cardiac repair with Postcon after 42 days of reperfusion, corroborated by attenuating degradation of ECM and deposition of newly synthesized collagens. Augmentation of the tissue repair with Postcon was still evident after its sparing effect on infarct size was blocked with 5-HD, suggesting multiple overlapping regulatory mechanisms involved in cardioprotection by Postcon. The protection achieved with Postcon was comparable to the benefits gained by Precon in all physiological endpoints measured. Our data were consistent with a recent study showing that repeated remote Postcon reduces adverse tissue remodeling and improves survival after myocardial infarction [56].
We have demonstrated that inhibition of apoptotic cell death with Postcon during early reperfusion is consistent with its attenuation of cytokine release [24]. In the present study, the sharp peak of plasma TNFα level was found at 30 min of reperfusion while IL-6 was persistently elevated during 3 h of reperfusion, potentially due to stimulation of reactive oxygen species and cytokine self-amplification pathways [23, 24]. In this regard, we have previously reported that Postcon inhibits superoxide radical generation [49] and lipid peroxidation [24] during reperfusion. To test whether activation of mitochondrial KATP channels is associated with the attenuation of cytokine production with Postcon, we selected 5-HD as a mitochondrial KATP channel blocker, and found that inhibition of TNFα and IL-6 with Postcon is blocked by 5-HD, consistent with the blockade of the sparing effect on infarct size with Postcon by 5-HD. Although we did not measure levels of cytokines during 42 days of reperfusion, we found that favorable tissue repair with Postcon is not altered by 5-HD, suggesting a lack of mitochondrial KATP channels in inhibition of adverse cardiac repair with Postcon.
It is generally agreed that the most important determinant of cardiac repair following infarction is the extent of the necrotic myocardium [21]. The adverse tissue repair due to extensive necrosis of the left ventricle mostly occurs, leading to scar formation and cardiac dysfunction [33]. Although ongoing experimental studies have consistently shown Postcon as a potential potent intervention for cardioprotection, it is unknown whether Postcon also plays a role in alleviating adverse tissue repair and improving cardiac function in addition to its sparing effect on infarct size. To demonstrate whether the beneficial effect of Postcon on cardiac repair is surely due to smaller infarct size, but not its substantial effect on tissue repair, we selected the 5-HD as a tool, which it has previously been shown to block the sparing effect of Postcon on infarct size [32], to test this hypothesis. Data presented clearly suggest that in the absence of reduction in infarct size with Postcon after 5-HD blockade, Postcon still has a favorable effect on tissue repair. If infarct size reduction is not only the effect of Postcon, therefore, we propose that it would be interesting to demonstrate whether the persistent beneficial effects also occur with Postcon that fails to reduce infarct size (i.e., a delayed Postcon maneuver). In this regard, recent studies have demonstrated that late focal cerebral and cardiac injury can be significantly attenuated, even when Postcon is applied in a delayed fashion, providing experimental evidence for long-term neuro- and cardioprotection with Postcon against ischemic/reperfused damage [27, 40, 42].
Healing of the infarcted tissue is a highly regulated process following the necrotic loss of cardiomyocytes. It begins with the activation of MMPs, a multigene family of zinc-dependent endopeptidases that are capable of degrading virtually every component of the ECM and resulting in formation of new fibrotic tissue in the infarcted myocardium [7]. Although the cell types that synthesize and secrete MMPs are not yet clear, it has been proposed that the stimulation of fibroblast-like cells, inflammatory cells, endothelial cells and cardiomyocytes by inflammatory mediators generated during reperfusion is capable of activating MMPs [12]. It has previously been reported that MMP activation starts early (less than 1 day after infarction) and is responsible for several aspects of infarct healing, including early ECM degradation, cell migration (inflammatory cells, fibroblasts), angiogenesis and remodeling of newly synthesized connective tissue [50]. As demonstrated in the present study, the pro-MMP-9 was not expressed and the pro-MMP-2 was generally found at low levels in normal and non-ischemic tissue. However, ischemia/reperfusion significantly up-regulated expression of active form MMP-2 at day 7. Although we do not know whether this is a peak in expression of myocardial MMP-2, this active form was still significantly detected at day 42, suggesting that MMP-2 may play a role in modulating ECM degradation during reperfusion [7]. Both pro-MMP-2 and MMP-2 were inhibited by Postcon and Precon during the course of the experiment, but not altered by 5-HD. These beneficial effects were consistent with reduced degradation of the original ECM identified by Masson’s trichrome staining and preservation of wall thickness analyzed by echocardiographic images.
Cardiac fibrosis is characterized by over-production of ECM, predominantly collagen types I and III, into the interstitial and perivascular space. Excessive collagen deposition leads to myocardial stiffening, impaired cardiac relaxation and filling, and overload of the heart [28, 51]. In the present study, we found a significant increase in levels of collagen types I and III at 7 and 42 days of reperfusion, indicating newly synthesized collagen content, which fits the time frame that the necrotic myocytes are entirely replaced by fibrous tissue [48]. Postcon and Precon comparatively reduced the amounts of collagen types I and III, consistent with the less collagen-rich wound healing as evidenced by Masson’s trichrome staining. Cells responsible for fibrous tissue formation after infarction consist principally of phenotypically transformed fibroblast-like cells, i.e., myofibroblasts, characterized by the expression of α-SMA microfilaments. On the site of the infarcted myocardium, myofibroblasts rapidly proliferate and accumulate, which are responsible for the scar formation via their expression of types I and III fibrillar collagens. Although we cannot outline the exact time window for collagen degradation and synthesis modulated by Postcon, the data in the present study support conclusions that Postcon may protect the ECM from degradation by attenuating MMPs during early phases of reperfusion and reduce the fibrotic tissue by constantly inhibiting collagen synthesis over time. Coincidence of the reduction in differentiation of fibroblasts to myofibroblasts and the level of tissue collagens by Postcon suggests myofibroblasts as a tissue extracollagen source.
The TGFβ1/Smad pathway plays a critical role in cardiac fibrosis via enhancing collagen synthesis [11, 58]. The members of the Smad family contain three distinct proteins: the TGFβ1 receptor-activated Smads (Smad1, Smad2, Smad3, Smad5 and Smad8), the mediator Smads (Smad4 and Smad10) and the inhibitory Smads (Smad6 and Smad7). Upon activation of the cellular surface TGFβ1 receptor, phosphorylated Smad2 and Smad3 form a complex with Smad4, translocate into the nucleus and subsequent bind to the TGFβ1-targeted collagen genes. The Smad6 and Smad7 function as inhibitors of TGFβ1 signaling by preventing Smad2/3 phosphorylation and disrupting Smad complex formation [6, 11, 58]. In the present study, we tested the hypothesis that inhibition of collagen with Postcon is mediated by TGFβ1/Smad signaling pathway. We found that changes in expression and phosphorylation of Smad2/3 and down-regulation of Smad7 were significantly detected at day 7, consistent with the time course in collagen synthesis during reperfusion. Given the fact that down-regulation of Smad7 is associated with enhanced expression of phosphorylated Smad2/3 [55], inhibition of phospho-Smad2/3 with Postcon was primarily associated with up-regulated Smad7. In the present study, the accumulation of α-SMA expressing myofibroblasts was significantly attenuated by Postcon and Precon in parallel with an inhibition in expression of TGFβ 1, indicating a role of TGFβ 1 in fibroblast-to-myofibroblast transformation during periods of fibrotic tissue formation [9]. Therefore, these data suggest that the favorable effects of Postcon on cardiac repair may largely be mediated by inhibiting collagen synthesis and fibrotic tissue formation through modulating TGFβ 1/Smad pathway.
Adequate infarct repair is suggested to be beneficial because it could prevent the most common complications that occur during the healing after myocardial injury, such as wall thinning, chamber dilatation and cardiac dysfunction [35, 41]. Current data confirm this hypothesis, because the promoted cardiac repair with Postcon may reduce wall thinning, attenuate lumen dilation, inhibit collagen deposition and further improve cardiac function. Recently, some of animal studies [10, 19, 39] and clinical observations [13, 45, 62] have failed to show a reduction in infarct size with Postcon. Potential explanations for the lack of a protective benefit from Postcon may include the animal models (i.e., healthy vs. co-morbidity models), the age and sex, the size of the area at risk myocardium, the time frame of infarction before starting treatment and the treatment interferes with other drugs [18, 62]. The present study was performed in a healthy animal model with blockade of the sparing effect on infarct size by Postcon. Data presented raise a possibility that the cardiac repair with Postcon could be achieved, even when the time window to reduce infarct size is missed. These data will also be of stimulus for clinical studies to perform retrospective studies on patients underwent Postcon without apparent clinical benefit, particularly for the patients treated with pharmacological intervention after myocardial infarction to show a long-term protection.
In summary, this is the first histological and functional study to provide experimental evidence showing the beneficial effect of Postcon on cardiac repair. We selected ECM degradation and collagen deposition as main markers, which have previously been used for identification of tissue repair [22, 61] to demonstrate protective effect of Postcon on cardiac healing after myocardial infarction. The results of the present study show that the promotion of cardiac repair could be achieved when the heart is Postcon by cycles of briefly interrupted perfusion during the early moments of reflow. To date, it has been shown that inflammatory cells [15], endothelial cells [59], cardiomyocytes [1] and fibroblasts (in the present study) could be postconditioned during reperfusion. Based on our previous reports [24, 59] and data obtained from the present study, we may conclude that Postcon reduces infarct size by inhibiting inflammatory response and cytokine production during early reperfusion, and promotes cardiac repair by reducing accumulation of myofibroblasts to balance ECM degradation and synthesis after extended reperfusion. However, more mechanistic studies are warranted to address whether activation of other signaling pathways, such as the reperfusion injury salvage kinase pathway, the survival activating factor enhancement pathway [16] or the Janus kinase-signal transducer and activator of transcription system, notably mitochondrial STAT3 pathway [20] is also involved in Postcon-modulated cardiac repair after myocardial infarction. Taken together, persistent promotion of cardiac repair with Postcon may represent a unique example for pharmacological treatment of myocardial infarction-derived heart failure.
References
Bell RM, Yellon DM (2012) Conditioning the whole heart—not just cardiomyocyte. J Mol Cell Cardiol 53:24–32. doi:10.1016/j.yjmcc.2012.04.001
Belvisi MG, Bottomley KM (2003) The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs? Inflamm Res 52:95–100. doi:http://www.ncbi.nlm.nih.gov/pubmed/12755372
Berthonneche C, Sulpice T, Boucher F, Gouraud L, de Leiris J, O’Connor SE, Herbert JM, Janiak P (2004) New insights into the pathological role of TNF-a in early cardiac dysfunction and subsequent heart failure after infarction in rats. Am J Physiol (Heart Circ Physiol) 287:H340–H350. doi:10.1152/ajpheart.01210.2003
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial SATAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. doi:10.1007/s00395-010-0124-1
Brown RD, Ambler SK, Mitchell MD, Long CS (2005) The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Ann Rev Pharmacol Toxicol 45:657–687. doi:10.1146/annurev.pharmtox.45.120403.095802
Bujak M, Frangogiannis NG (2006) The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovascular Res 74:184–195. doi:10.1016/j.cardiores.2006.10.002
Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R (2000) Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 101:1833–1839. doi:10.1161/01.CIR.101.15.1833
Cohen MV, Yang XM, Downey JM (2007) The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 115:1895–1903. doi:10.1161/CIRCULATIONAHA.106.675710
Dobazewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG (2010) Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 107:418–428. doi:10.1161/CIRCRESAHA.109.216101
Douhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B (2008) Myocardial postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol (Heart Circ Physiol) 295:H1508–H1586. doi:10.1152/ajpheart.00379.2008
Euler-Taimor G, Heger J (2006) The complex pattern of SMAD signaling in the cardiovascular system. Cardiovascular Res 69:15–25. doi:10.1016/j.cardiores.2005.07.007
Falk V, Soccal PM, Grunenfelder J, Hoyt G, Walther T, Robbins RC (2002) Regulation of matrix metalloproteinases and effect of MMP-inhibition in heart transplant related reperfusion injury. Eur J Cardio thorac Surg 22:53–58. doi:10.1007/s00109-004-0606-4
Freixa X, Bellera N, Ortiz-Perez JT, Jimenez M, Pare C, Bosch X, De Caralt TM, Betriu A, Masotti M (2012) Ischemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J 33:103–112. doi:10.1093/eurheartj/ehr297
Garcia S, Henry TD, Wang YL, Chavez IJ, Pedersen WR, Lesser JR, Shroff GR, Moore L, Traverse JH (2011) Long-term follow-up of patients underlying postconditioning during ST-elevation myocardial infarction. J Cardiovasc Trans Res 4:92–98. doi:10.1007/s12265-010-9252-0
Granfeldt AV-J, Jiang R, Wang NP, Mykytenko J, Eldaif S, Deneve J, Guyton RA, Zhao ZQ, Vinten-Johansen J (2012) Neutrophil inhibition contributes to cardioprotection by postconditioning. Acta Anaesthesiol Scand 56:48–56. doi:10.1111/j.1399-6576.2011.02577.x
Hausenloy DJ, Lecour S, Yellon DM (2011) RISK and SAFE pro-survival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal 14:893–907. doi:10.1089/ars.2010.3360
Heinzel FR, Luo Y, Li XK, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G (2005) Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Cir Res 97:583–586. doi:10.1161/01.RES.0000181171.65293.65
Heusch G (2011) Reduction of infarct size by ischemic post-conditioning in humans: fact or fiction? Eur Heart J 33:13–15. doi:10.1093/eurheartj/ehr341
Heusch G, Bu chert A, Feldhaus S, Schulz R (2006) No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res Cardiol 101:354–356. doi:10.1007/s00395-006-0589-0
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. doi:10.1161/CIRCRESAHA.111.255604
Heusch G (2012) Cardioprotection: chances and challenges of its translation to the clinic. Lancet. doi: 10.1016/S0140-6736(12)60916-7
Hinz B (2007) Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127:526–537. doi:10.1038/sj.jid.5700613
Kleinbongard P, Heusch G, Schulz R (2010) TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Therap 127:295–314. doi:10.1016/j.pharmthera.2010.05.002
Kin H, Wang NP, Mykytenko J, Reeves J, Deneve J, Jiang R, Zatta AJ, Guyton RA, Vinten-Johansen J, Zhao ZQ (2007) Inhibition of myocardial apoptosis by postconditioning is associated with attenuation of oxidative stress-mediated nuclear factor-kappa B translocation and TNF alpha release. Shock 29:761–768. doi:10.1097/SHK.0b013e31815cfd5a
Lonborg J, Kelbak H, Vejlstrup N, Jorgensen E, Helqvist S, Saunamaki K, Clemmensen P, Holmvang L, Treiman M, Jensen JS, Engstrom T (2010) Cardioprotective effects of ischemic postconditioning in patients treated with primary percutaneous coronary intervention, evaluated by magnetic resonance. Circ Cardiovasc Interv 3:34–41. doi:10.1161/CIRCINTERVENTIONS.109.905521
Lonborg J, Holmvang L, Kelbak H, Vejlstrup N, Jorgensen E, Helqvist S, Saunamaki K, Clemmensen P, Treiman M, Jensen JS, Engstrom T (2010) ST-segment resolution and clinical outcome with ischemic postconditioning and comparison to magnetic resonance. Am Heart J 160:1085–1091. doi:10.1016/j.ahj.2010.09.026
Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M (2009) Delayed postconditioning protects against cerebral ischemia in the mouse. Stroke 40:3349–3355. doi:10.1161/STROKEAHA.109.557314
Lindsey ML, Mann DL, Entman ML, Spinale FG (2003) Extracellular matrix remodeling following myocardial injury. Ann Med 35:316–326. doi:10.1080/07853890310001285
Longacre LS, Kloner RA, Arai AE, Baners CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, Jennings RB, Lefer DJ, Mentzer RM, Murphy E, Ovize M, Ping P, Przyklenk K, Sack MN, Vander Heider RS, Vinten-Johansen J, Yellon DM (2011) New horizons in cardioprotection: recommendations from the 2010 national heart, lung, and blood insititute workshop. Circulation 124:1172–1179. doi:10.1161/CIRCULATIONAHA.111.032698
Luo W, Li B, Lin G, Huang R (2007) Postconditioning in cardiac surgery for tetralogy of Fallot. J Thorac Cardiovasc Surg 133:1373–1374. doi:10.1016/j.jtcvs.2007.01.028
Minatoguchi S, Takemura G, Chen XH, Wang N, Uno Y, Koda M, Arai M, Misao Y, Lu C, Suzuki K, Goto K, Komada A, Takahashi T, Kosai K, Fujiwara T, Fujiwara H (2004) Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation 109:2572–2580. doi:10.1161/01.CIR.0000129770.93985.3E
Mykytenko J, Reeves JG, Kin H, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao ZQ (2008) Persistent beneficial effect of postconditioning against infarct size: role of mitochondrial KATP channel activation during reperfusion. Basic Res Cardiol 103:472–484. doi:10.1007/s00395-008-0731-2
Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, Minatoguchi S, Fujiwara T, Fujiwara H (2005) Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation 111:2430–2437. doi:10.1161/01.CIR.0000165066.71481.8E
Pain T, Yang X-M, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM (2000) Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res 87:460–466. doi:10.1161/01.RES.87.6.460
Payne TR, Oshima H, Okada M, Momoi N, Tobita K, Keller BB, Peng H, Huard J (2007) A relationship between vascular endothelial growth factor, angiogenesis, and cardiac repair after muscle stem cell transplantation into ischemic hearts. J Am Coll Cardiol 50:1677–1684. doi:10.1016/j.jacc.2007.04.100
Penna C, Pasqua T, Perrelli MG, Pagliaro P, Cerra MC, Angelone T (2012) Postconditioning with glucagon like peptide-2 reduces ischemia-reperfusion injury in isolated rat hearts: role of survival kinases and mitochondrial KATP channels. Basic Res Cardiol 107:272–280. doi:10.1007/s00395-012-0272-6
Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P (2006) Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol 101:180–189. doi:10.1007/s00395-006-0584-5
Prasad A, Stone GW, Holmes DR, Gersh B, Dphil C (2009) Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation 120:2105–2112. doi:10.1161/CIRCULATIONAHA.108.814640
Przyklenk K, Maynard M, Greiner DL, Whittaker P (2011) Cardioprotection with postconditioning: loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid Redox Signal 14:781–790. doi:10.1089/ars.2010.3343
Ren C, Gao XW, Niu G, Yan ZM, Chen XY, Zhao H (2008) Delayed postconditioning protects against focal ischemic brain injury in rats. PLoS ONE 3:1–12. doi:10.1371/journal.pone.0003851
Rohde LE, Aikawa M, Cheng GC, Sukhova G, Solomon SD, Libby P, Pfeffer J, Pfeffer MA, Lee RT (1999) Echocardiography-derived left ventricular end-systolic regional wall stress and matrix remodeling after experimental myocardial infarction. J Am Coll Cardiol 33:835–842. doi:10.1016/S0735-1097(98)00602-0
Roubille F, Franck-Miclo A, Covinhes A, Lafont C, Cransac F, Combes S, Vincent A, Fontanaud P, Sportouch-Dukhan C, Redt-Clouet C, Nargeot J, Piot C, Barre’re-Lemaire S (2011) Delayed postconditioning in the mouse heart in vivo. Circulation 124:1330–1336. doi:10.1161/CIRCULATIONAHA.111.031864
Sadat U, Walsh SR, Varty K (2008) Cardioprotection by ischemic postconditioning during surgical procedures. Expert Rev Cardiovasc Ther 6:999–1006. doi:10.1586/14779072.6.7.999
Sandu N, Schaller B (2010) Postconditioning: a new or old option after ischemic stroke? Expert Rev 8:479–482. doi:10.1586/erc.09.180
Sorensson P, Salem N, Bouvier F, Bohm F, Settergren M, Caidahl K, Tornvall P, Arheden H, Ryden L, Pernow J (2010) Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart 96:1710–1715. doi:10.1136/hrt.2010.199430
Spinale FG, Gunasinghe H, Sprunger PD, Baskin JM, Bradham WC (2002) Extracellular degradative pathways in myocardial remodeling and progression to heart failure. J Card Fail 8:S332–S338. doi:10.1054/jcaf.2002.129259
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit J-F, Bonnefoy E, Finet G, Andre-Fouet X, Ovize M (2005) Postconditioning the human heart. Circulation 112:2143–2148. doi:10.1161/CIRCULATIONAHA.105.558122
Sun Y (2009) Myocardial repair/remodeling following infarction: roles of local factors. Cardiovascular Res 81:482–490. doi:10.1016/j.yjmcc.2009.08.002
Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ (2005) Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2 + overload. Am J Physiol (Heart Circ Physiol) 288:H1900–H1908. doi:10.1152/ajpheart.01244.2003
Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP (2004) Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci 74:1561–1572. doi:10.1016/j.lfs.2003.09.042
Tessone A, Feinberg MS, Barbash IM, Reich R, Holbova R, Richmann M, Mardor Y, Leor J (2005) Effect of matrix metalloproteinase inhibition by doxycycline on myocardial healing and remodeling after myocardial infarction. Cardiovasc Drugs Ther 19:383–390. doi:10.1007/s10557-005-5201-6
Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, Cung TT, Bonnefoy E, Angoulvant D, Aupetit JF, Finet G, Andre-Fouet X, Macia JC, Raczka F, Rossi R, Itti R, Kirkorian G, Derumeaux G, Ovize M (2008) Long-term benefit of postconditioning. Circulation 117:1037–1044. doi:10.1161/CIRCULATIONAHA.107.729780
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM (2004) Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res 95:230–232. doi:10.1161/01.RES.0000138303.76488.fe
Vinten-Johansen J, Zhao ZQ, Zatta AJ, Kin H, Halkos ME, Kerendi F (2005) Postconditioning—A new link in nature’s armor against myocardial ischemia-reperfusion injury. Basic Res Cardiol 100:295–310. doi:10.1007/s00395-005-0523-x
Wang B, Omara, Angelovska T, Drobic V, Rattan SG, Jones SC, Dixon IMC (2007) Regulation of collagen synthesis by inhibitory Smad7 in cardiac myofibroblasts. Am J Physiol 293:H1282–H1290. doi:10.1152/ajpheart.00910.2006
Wei M, Xin P, Li SA, Tao JP, Li YP, Li J, Liu MY, Li JB, Zhu W, Redington AN (2011) Repeated remote ischemic postconditioning protects against adverse left ventricular remodeling and improves survival in a rat model of myocardial infarction. Cir Res 108:1220–1225. doi:10.1161/CIRCRESAHA.110.236190
Yang XC, Liu Y, Wang LF, Cui L, Wang T, Ge YG, Wang HS, Li WM, Xu L, Ni ZH, Liu SH, Zhang L, Jia HM, Vinten-Johansen J, Zhao ZQ (2007) Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J Invasive Cardiol 19:424–430
Yuan SM, Jing H (2010) Cardiac pathologies in relation to Smad-dependent pathways. Interact Cardiovasc Thorac Surg 11:455–460. doi:10.1510/icvts.2010.234773
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol (Heart Circ Physiol) 285:H579–H588. doi:10.1152/ajpheart.01064.2002
Zhao ZQ, Nakamura M, Wang N-P, Velez DA, Hewan-Lowe KO, Guyton RA, Vinten-Johansen J (2000) Dynamic progression of contractile and endothelial dysfunction and infarct extension in the late phase of reperfusion. J Surg Res 94:1–12. doi:10.1006/jsre.2000.6029
Zhao ZQ, Puskas JD, Xu D, Wang N-P, Guyton RA, Vinten-Johansen J, Matheny R (2008) Improvement in cardiac function with small intestine extracellular matrix is associated with recruitment of c-kit cells, myofibroblasts, and macrophages after myocardial infarction. J Am Coll Cardiol 55:1250–1261. doi:10.1016/j.jacc.2009.10.049
Zhou C, Li L (2012) Age may contribute to the negative cardiac effect of postconditioning on STEMI patients. Int J Cardiol. doi: 10.1016/j.ijcard.2012.09.174
Acknowledgments
This study was supported in part by a seed Grant from the Mercer University School of Medicine and National Natural Science Foundation of China (81170145/H0203).
Conflict of interest
No conflicts of interest were declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Z.-F. Wang and N.-P. Wang contributed equally to this work.
The article “Postconditioning promotes the cardiac repair through balancing collagen degradation and synthesis after myocardial infarction in rats”, Basic Res Cardiol (2013) 108:318, was retracted by the authors who regret to have used different fields of the same samples for MMP staining and western blot assay to represent 2 distinct groups on 2 occasions in Figure 4 and 5. Some of the raw data for the earlier experiments with the use of echocardiography for Figure 8 and 9A were not available for further analysis to exclude that also the same samples were used for 2 distinct groups. The authors regret the effect of this action on the work of other investigators.
An erratum to this article is available at http://dx.doi.org/10.1007/s00395-015-0469-6.
About this article
Cite this article
Wang, ZF., Wang, NP., Harmouche, S. et al. RETRACTED ARTICLE: Postconditioning promotes the cardiac repair through balancing collagen degradation and synthesis after myocardial infarction in rats. Basic Res Cardiol 108, 318 (2013). https://doi.org/10.1007/s00395-012-0318-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0318-9