
Abstract
G-CSF (granulocyte colony-stimulating factor) treatment has been shown to cause beneficial effects including a reduction of inducible arrhythmias in rodent models of ischemic cardiomyopathy. The aim of the present study was to test whether these effects do also apply to pacing-induced non-ischemic heart failure. In 24 female rabbits, heart failure was induced by rapid ventricular pacing. 24 rabbits were sham operated. The paced rabbits developed a significant decrease of ejection fraction. 11 heart failure rabbits (CHF) and 11 sham-operated (S) rabbits served as controls, whereas 13 sham (S-G-CSF) and 13 heart failure rabbits (CHF-G-CSF) were treated with 10 μg/kg G-CSF s.c. over 17 ± 4 days. G-CSF treatment caused a ~25% increased arterial and capillary density and a ~60% increased connexin 43 expression in failing hearts. In isolated, Langendorff-perfused rabbit hearts eight monophasic action potential recordings showed prolongation of repolarization in CHF as compared with controls in the presence of the QT prolonging agent erythromycin (+33 ± 12 ms; p < 0.01). Moreover, a significant increase in dispersion of repolarization contributed to a significantly higher rate of ventricular tachyarrhythmias in CHF. G-CSF-pre-treated hearts showed a further increase in prolongation of repolarization as compared with S and CHF. The further increase in dispersion of repolarization [S-G-CSF: +23 ± 9 ms (spatial), +13 ± 7 ms (temporal); CHF-G-CSF: +38 ± 14 ms (spatial), +10 ± 4 ms (temporal); p < 0.05 as compared with S and CHF], increased the incidence of ventricular tachyarrhythmias. In summary, chronic G-CSF treatment has moderate beneficial effects on parameters potentially related to hemodynamic function in the non-ischemic rabbit CHF model. However, a significant reduction of repolarization reserve might seriously challenge its suitability as a therapeutic agent for chronic CHF therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic heart failure (CHF) is a common clinical problem and the final manifestation of many cardiovascular disorders. Patients with CHF have a 6–9 times higher rate of sudden cardiac death as compared with the general population and up to 50% of deaths in patients with CHF are sudden. The majority of these are due to ventricular tachyarrhythmias [46]. Numerous discrepancies regarding the ionic and molecular changes in CHF have been reported. However, a consistent finding is prolongation of cardiac repolarization that results in a reduced repolarization reserve. This concept was first introduced by Dan Roden [44], implying that several different circumstances influence the repolarization process and facilitate the occurrence of life-threatening ventricular tachyarrhythmias. In CHF, repolarization reserve may be further diminished by repolarization prolonging drugs, especially blockers of the rapid component of the delayed rectifier potassium current I Kr [16]. Furthermore, conditions such as hypomagnesemia, hypokalemia, bradycardia, drug accumulation due to liver or renal failure [1], or inhibition of cytochrome P450 drug metabolism [43] enhance proarrhythmia in CHF.
Several groups [11, 15, 41, 56] including our own [30] showed that granulocyte colony-stimulating factor (G-CSF) treatment improved hemodynamic function and potentially underlying structural parameters such as arterial density in animal models of ischemic heart disease (for review: [28, 49]) as it had also been demonstrated for other growth factors such as FGF-2 and VEGF (for review: [32]). We demonstrated that it also led to a reduction of induced ventricular tachycardias in this model [30]. In non-ischemic models, including doxorubicin [34] as well as angiotensin II-induced heart failure [27], G-CSF application was also shown to exert beneficial effects. However, whether G-CSF therapy diminishes proarrhythmia in such chronic non-ischemic heart failure models has not been investigated so far.
Accordingly, the aims of the present study were to: (1) test whether G-CSF treatment of rabbit hearts affected by non-ischemic heart failure influences the incidence of ventricular tachyarrhythmias and, if this applies, to (2) comparatively investigate presumably underlying electrical parameters such as repolarization and its spatial and temporal dispersion. The corresponding analyses should then also include an experimental reduction of the repolarization reserve using the I Kr-blocking agent erythromycin to simulate the presence of repolarization prolonging drugs in CHF potentially potentiating possible effects of G-CSF. (3) Finally, the arterial and capillary density should be analyzed to assess effects of the cytokine on the non-ischemic failing heart potentially relevant for cardiac pump function.
Methods
All experimental protocols were approved by the local animal care committee and conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Induction of heart failure and administration of G-CSF
CHF was induced in 24 rabbits via 3–4 weeks of ventricular pacing at 400 beats/min. The method of induction of CHF has recently been reported in detail by others [19, 20, 52] and our group [14, 35]. In brief, adult female New Zealand white rabbits were anesthetized with ketamine (75 mg/kg) and xylazine (5.8 mg/kg). A pacemaker lead was implanted into the right ventricle and started stimulating with 400 beats/min 1 week later. Over a period of 3–4 weeks the rabbits were monitored by clinical examination and echocardiography (Fig. 1a). In addition, 24 sham-operated rabbits were observed over the same period. 11 sham-operated rabbits (S) and 11 heart failure (CHF) rabbits were treated with saline and served as controls, whereas 13 sham-operated rabbits (S-G-CSF) and 13 heart failure rabbits (CHF-G-CSF) were treated with recombinant human G-CSF (Granocyte, Chugai Pharma Marketing Ltd.; 10 μg/kg/day) s.c. over a period of 17 ± 4 days.

a Flow chart that illustrates the time-line of events from pacemaker implant to histopathology examinations. b Flow chart illustrating the different consecutive parts of the experimental protocol
Preparation of hearts for perfusion
The method of preparing the hearts has previously been described in detail [37, 38]. Female New Zealand white rabbits (n = 48) weighing 3.5–4.0 kg were anesthetized with sodium thiopental (200–300 mg i.v.). After midsternal incision and opening of the pericardium, the complete hearts were removed and immediately placed in an ice-cold Krebs–Henseleit solution (composition in mM: CaCl2 1.80, KCl 4.70, KH2PO4 1.18, MgSO4 0.83, NaCl 118, NaHCO3 24.88, Na-pyruvate 2.0 and d-glucose 5.55). The spontaneously beating hearts were perfused at constant flow (52 ml/min) with warm (36.8–37.2°C) Krebs–Henseleit solution. The cannulated and perfused hearts were attached to a vertical Langendorff apparatus (Hugo Sachs Elektronic, Medical Research Instrumentation, March-Hugstetten, Germany). A deflated latex balloon was inserted into the left ventricle and connected to a pressure transducer to control haemodynamic stability. The atrioventricular (AV) node was ablated to slow the intrinsic heart rate.
Histopathology and immunofluorescence
Tissue samples of Langendorff-perfused hearts were fixed in 3.7% formaldehyde, followed by dehydration in 10% sucrose solution, OCT embedding, and cryoconservation for azan staining (standard procedure) or immunofluorescent detection as described [30]. The following primary antibodies were used: anti connexin 43 (Sigma-Aldrich Inc, USA, Cat.-Nr. C-6219—polyclonal antibody); anti-alpha smooth muscle actin (Abcam, USA, Cat.-Nr. ab24115). Secondary antibodies were biotinylated and were finally labeled by a streptavidin-coupled fluorescent dye (Alexa Fluor 594 streptavidin; Invitrogen), or the immunohistochemical signal was further amplified after incubation with the secondary antibody using the TSA Fluorescence System with Cyanine 3 (Cy3) as the final label (NEL704A; PerkinElmer). For the simultaneous detection of capillaries and arteries the anti-alpha smooth muscle actin antibody was used in combination with TRITC-labeled BS1 lectin (Sigma-Aldrich), which is known to specifically bind to endothelial cells. For quantification arterial vessels were counted in 15 optical fields representing 15 independent tissue sections per animal. The summarized numbers of arterial vessels per animal were used for the calculation of the mean values of arterial density in animals of the various experimental groups. Capillaries were counted in 12 optical fields per animal and the average number of capillaries per optical field and animal was calculated. The values obtained for single animals were used for the calculation of mean capillary densities.
The quantification of connexin 43 immunofluorescence signals was done as described [30] using the public domain software Image J.
Hydroxyproline and cardiac troponin I detection
The 4-hydroxyproline content of cardiac tissue samples was determined as previously reported [40]. Briefly, proteins in tissue samples were hydrolyzed into amino acids in 6 M HCl. The amino acids were transformed to phenylthiocarbamoyl (PTC) derivatives and separated by HPLC. Amino acid standards were used for quantification. Analysis was done in duplicate for each sample.
Cardiac troponin I was detected by western blotting of 50 µg total protein homogenate per lane according to a standard protocol [55] using a monoclonal anti-cardiac troponin I antibody (Abcam, USA, Cat.# ab55819) in combination with a horseradish peroxidase-coupled anti-mouse secondary antibody (Amersham Biosciences, Germany, Cat.-No. NA931), the ECL chemiluminescence system (ECL plus, Amersham Bioscience, Germany) and densitometry of the X-ray films (Hyperfilm, Amersham Bioscience, Germany). Densitometric scans were quantified using ImageQuant imaging software (Molecular Dynamics, Sunnyvale, USA).
Electrocardiographic and electrophysiologic measurements
A volume-conducted ECG was recorded by complete immersion of the heart into a bath of Krebs–Henseleit solution. Signals were amplified by a standard ECG amplifier (filter settings: 0.1–300 Hz). Monophasic action potential (MAP) recordings and stimulation were accomplished using contact MAP pacing catheters (EP Technologies, Mountain View, CA, USA). The MAP electrograms were amplified and filtered (low pass 0.1 Hz, high pass 300 Hz). The recordings were considered reproducible and, therefore, acceptable for analysis only if they had a stable baseline amplitude with a variation of less than 20% for at least 60 s during each cycle length and a stable duration with a variation of less than 10% in this time window measured at 90% repolarization (MAP90). Seven MAPs were evenly spread in a circular pattern around both ventricles, one MAP was recorded from the left endocardium. Pacing at twice diastolic threshold was performed for 1 min at each cycle length (CL) from 900–300 ms using a programmable stimulator (Universal Programmable Stimulator, UHS 20, Biotronik, Germany) which delivered square-wave pulses of 2 ms pulse width. All data were digitized at a rate of 1 kHz with 12-bit resolution and subsequently stored on a removable hard disk (BARD LabSystem, Bard Electrophysiology, Murray Hill, Massachusetts, USA).
Experimental protocol
After placing the MAP catheters and inducing complete AV block, CL-dependence was first investigated under baseline conditions by pacing the hearts at cycle lengths between 900 and 300 ms. Thereafter, the I Kr-blocking agent erythromycin (300 μM) was infused just above the heart [12] (Fig. 1b). We choose erythromycin as a strong I Kr blocker because of a long experience in our lab [36] and that of other groups [47]. The concentration of erythromycin was higher than the expected free plasma concentration to create a proarrhythmic milieu and to better study the underlying mechanisms of arrhythmogenesis in heart failure.
Pacing, MAP recording and measurement of ECG parameters were started 20 min after drug infusion. The potassium concentration was lowered to 1.5 mM to provoke early afterdepolarizations (EAD) which are defined as a positive voltage deflection that interrupted the smooth contour of phase 2 or 3 repolarization of the action potential and ventricular tachyarrhythmias in spontaneously beating bradycardic hearts. Low potassium concentration has been demonstrated to exert additional block of I Kr, even in the presence of maximal drug-related I Kr block [64]. MAP90 was measured as the average interval between the fastest MAP upstroke and 90% repolarization. Dispersion of MAP90 was expressed as the difference between the minimum and the maximum of MAP90, simultaneously recorded in eight endocardial and epicardial catheters. Since activation time is negligible in this animal model, dispersion of MAP90 is treated as equivalent to dispersion of repolarization. Temporal dispersion was determined in spontaneously beating hearts by analyzing 30 consecutive beats through Poincaré plots by plotting each MAP against the former MAP [24, 62]. Measurements were performed after 20 min of drug infusion directly before the pacing protocol. At this time a steady state was achieved with a very stable ventricular escape rhythm between 50 and 60 beats per minute. There was no dispersion of intrinsic cycle. The areas of the plots were determined and their dimensions calculated. The mean orthogonal distance from the diagonal to the points of the Poincaré plot was determined and referred to as temporal dispersion (STV = |D n + 1 − D n |/[30 × 2]), where D represents the duration of APD90. This nomenclature is adopted from investigations using long-term Holter monitoring of humans, in which steady changes tend to follow the diagonal and sudden changes (e.g., an extrasystole) result in a deviation from the diagonal [5].
Data acquisition and statistical analysis
The observed data were entered onto a computerized database (Microsoft Excel2003). Statistical analysis was performed using the SPSS Software, version 18.0.0. (07/30/2009; SPSS, Inc., Chicago, USA) or SigmaStat, version 3.1 (Systat Software Inc., San Jose, CA, USA). Categorical variables are expressed as frequency and percentage, whereas continuous variables are presented as mean ± SD. Before statistical testing, each continuous variable was analyzed for its normal distribution using the Kolmogorov–Smirnov test. The nonparametric dependent variables (cycle length influence of erythromycin and G-CSF-pre-treatment on ECG parameters, MAP duration, spatial dispersion and temporal dispersion of repolarization) were assessed using the Friedman test. The nonparametric independent variable was assessed using the Kruskal–Wallis test. Pairwise multiple comparisons following the Friedman and Kruskal–Wallis tests, respectively, were performed using the procedure proposed by Dunn. The McNemar’s test was used to compare the rate of inducibility of EADs and TdP after infusion of erythromycin. The Chi-squared test was used to compare the occurrence of EAD and TdP, respectively, between study groups. For the statistical analysis of hydroxyproline values, vessel densities and connexin 43 expression, an unpaired t test was applied by the SigmaStat software in cases of normal distribution and equal variance. Otherwise the Mann–Whitney Rank Sum Test was used. Differences were considered significant at p < 0.05.
Results
Assessment of heart failure and structural effects of G-CSF potentially related to cardiac pump function
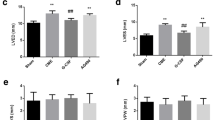
During the period of fast ventricular pacing, rabbits became lethargic, fatigued, showed loss of appetite and developed respiratory distress. Moreover, the paced rabbits developed ascites, pleural and pericardial effusion and peripheral edema [14]. Two-dimensional echocardiography demonstrated a significant decrease in ejection fraction (EF) from 68 ± 7 to 18 ± 5% in controls and from 71 ± 8 to 17 ± 4% in G-CSF treated hearts in the presence of a dilated left ventricle and significantly enlarged atria after 3–4 weeks of pacing. These findings are in accordance with previous results in the same model [14]. There was a trend to later development of an EF below 20% (19.6 ± 4.1 days in G-CSF treated animals vs. 16.7 ± 4.7 days in untreated hearts). Rabbit hearts developed marked interstitial fibrosis during pacing as revealed by azan staining (not shown). The quantitative assessment of hydroxyproline contents as a marker for collagen (Fig. 2a) as well as western blotting using an anti-cardiac troponin I antibody (Fig. 2b) suggested a slight trend towards a reduction of fibrosis in G-CSF treated failing hearts as compared to controls. In addition, arterial and capillary densities were assessed in hearts of the four experimental animal groups. G-CSF treatment caused a significant increase of both, arterial and capillary density, in control and paced rabbit hearts as compared with untreated hearts (Fig. 3).

a Hydroxyproline content (nmoles/mg cardiac tissue) of failing and sham hearts untreated or treated with G-CSF. Assuming that pacing could only increase the hydroxyproline content single-sided t test-based statistical analysis was applied and revealed significantly different mean hydroxyproline values for sham and paced rabbit hearts without G-CSF treatment (p = 0.029). N numbers of hearts investigated. b Expression of cardiac troponin T in failing hearts and hearts from sham-operated animals untreated or treated with G-CSF as assessed by western blotting with an anti-cardiac troponin T antibody

Influence of G-CSF on arterial and capillary density in sham- and CHF-rabbit left ventricle. a Representative fluorescence-based detection of arteries (green immunofluorescence of α-smooth muscle actin) and capillaries (red fluorescence caused by BS1 lectin binding to endothelial cells) in a paced rabbit heart. Magnification ×200. Mean numbers of arterial vessels (b) and capillaries (c) in LV sections of rabbit hearts (n numbers of animals investigated) treated as indicated
Effect of G-CSF therapy on action potential duration in sham and failing hearts in the presence of erythromycin
During electrophysiologic examination, all electrocardiographic parameters reached equilibrium within 10 min. In non-failing hearts, not treated with erythromycin, there was no difference in MAP duration between G-CSF and untreated hearts (Fig. 4a). After developing CHF, thus reducing repolarization reserve both curves [hearts treated with G-CSF (closed rhombus) and hearts not treated with G-CSF (open rhombus)] separated from each other (Fig. 4b). Adding erythromycin-related I Kr block as another contributing factor of a reduced repolarization reserve led to a further increase in action potential duration in sham (Fig. 4c) and especially in failing hearts (Fig. 4d). Failing hearts (not treated with G-CSF) showed a significant prolongation of repolarization (+33 ± 12 ms; p < 0.01) at cycle lengths between 900 and 300 ms in the presence of erythromycin as compared with sham (erythromycin-treated) hearts. G-CSF administration led to a further significant increase in MAP90 in sham hearts (S-G-CSF: +17 ± 6 ms; p < 0.05 as compared with S hearts (except for stimulation cycle lengths of 300 and 400 ms; Fig. 4c) and in failing hearts (CHF-G-CSF: +44 ± 16 ms; p < 0.05 as compared with CHF; Fig. 4d) thereby presenting a third factor that results in a reduction of myocardial repolarization reserve. The increase in action potential duration was accompanied by a significant increase in QT interval.

G-CSF effect on action potential duration in sham-operated hearts as compared to CHF hearts, under baseline conditions and after administration of 300 μM erythromycin: open diamond untreated hearts, closed diamond G-CSF-treated hearts; closed star p < 0.05. a Almost congruent curves in hearts, not treated with erythromycin and without prior induction of CHF, closed rhombus treated with G-CSF, open rhombus not treated with G-CSF. b Hearts not treated with erythromycin, after developing CHF, closed rhombus treated with G-CSF, open rhombus not treated with G-CSF. c Hearts treated with erythromycin without prior induction of CHF, closed rhombus treated with G-CSF, open rhombus not treated with G-CSF. d Peak deviation of both curves after implementing all factors that reduce repolarization reserve: hearts treated with erythromycin after induction of CHF, closed rhombus treated with G-CSF, open rhombus not treated with G-CSF
In the presence of 300 μM erythromycin, the increase in MAP duration ranged between 11% at a cycle length of 300 ms and 30% at a cycle length of 900 ms (p < 0.05) in failing hearts without G-CSF-treatment. This marked reverse-frequency dependence was even more prominent in failing hearts previously treated with G-CSF (mean increase 21% at 300 ms and 44% at 900 ms, p < 0.05; Fig. 4) and could also be observed in sham hearts treated with G-CSF (mean increase 12% at 300 ms and 36% at 900 compared to 6% and 12% in hearts without pre-treatment; Fig. 4).
Spatial and temporal dispersion of repolarization
In the presence of erythromycin, MAPs demonstrated a slight but significant increase in spatial dispersion of repolarization in heart failure as compared with controls (S = 46 ± 16 ms to CHF = 55 ± 19 ms; p < 0.05; Fig. 5). Administration of G-CSF led to a further increase in dispersion of repolarization. In sham-operated hearts an increase in total spatial dispersion of repolarization of 23 ms (S = 46 ± 16 ms to S-G-CSF = 69 ± 22 ms; p = 0.001; Fig. 5) occurred. In CHF, G-CSF led to an increase of total dispersion of 38 ± 14 ms (HF: 55 ± 19 ms to CHF-G-CSF: 93 ± 24 ms; p < 0.05; Fig. 5).

Significant increase in total dispersion of repolarization in the presence of erythromycin administration in CHF as compared with sham hearts. G-CSF therapy led to a further relevant increase in dispersion in sham hearts and in CHF hearts
In untreated failing hearts (10.1 ± 1.3 ms), temporal dispersion did not differ significantly from values of control hearts (9.7 ± 2 ms; p = ns). Application of erythromycin did not cause significant changes in sham hearts but elevated temporal dispersion to 18.4 ± 3.7 ms (p < 0.05) in failing hearts. In the presence of erythromycin, control hearts pre-treated with G-CSF showed a more marked increase of temporal dispersion of repolarization (+13 ± 7 ms; p < 0.05; Fig. 6, left bottom) as compared to controls, not treated with G-CSF (Fig. 6, left top) after application of erythromycin. Moreover, G-CSF pre-treatment led to a further significant increase in temporal dispersion of repolarization in heart failure (Fig. 6, right bottom; p < 0.05) as compared to controls. The increase in spatial and temporal dispersion of repolarization is comparable to the increase of action potential duration with a more marked effect the more repolarization reserve is affected (erythromycin + CHF + G-CSF).

Representative example of temporal dispersion of repolarization displayed in Poincaré plots. Top left sham hearts without G-CSF, baseline and after administration of 300 μM erythromycin. Top right after induction of CHF with a significant increase in the presence of erythromycin. Bottom left sham hearts treated with G-CSF. Bottom right pretreated with G-CSF, after induction of CHF with a significant increase in the presence of erythromycin
Early afterdepolarizations and ventricular tachyarrhythmias
After complete AV block and lowering of potassium concentration, 5 of 11 sham (S) and 8 of 11 failing hearts showed EAD and ventricular tachyarrhythmias in the presence of erythromycin (Fig. 7a, left). In 13 G-CSF-pre-treated hearts ventricular tachyarrhythmias were seen in 8 (S-G-CSF) and 11 (CHF-G-CSF) hearts (Fig. 7a, right). They were always associated with EAD (p < 0.05; McNemar test). In the presence of erythromycin, CHF also significantly increased the number of ventricular tachyarrhythmias (CHF = 127 single episodes vs. S = 27 single episodes; p = 0.045). G-CSF caused a further increase in their incidence (Fig. 7b, c).

a Number of hearts with ventricular tachyarrhythmias (%) in the presence of 300 μM erythromycin in untreated rabbits (left n = 11) and after therapy with G-CSF (right n = 13) with a significant increase in CHF as compared with sham hearts for both groups and a trend towards more arrhythmias after pretreatment with G-CSF. b Single episodes of ventricular tachyarrhythmias with significantly more episodes in CHF as compared to sham hearts and more episodes in G-CSF hearts as compared to both (sham and CHF) untreated groups. c Typical example of a polymorphic ventricular tachyarrhythmia in the presence of 300 μM erythromycin in a rabbit failing heart, pretreated with G-CSF during Langendorff perfusion
G-CSF influence on connexin 43 expression
The expression of connexin 43 was investigated in situ using immunofluorescence. A statistically significant reduction of connexin 43 was detected in CHF hearts, as compared to control (Fig. 8). G-CSF treatment caused a significant increase of connexin 43 expression in failing hearts (CHF vs. CHF-G-CSF, p = 0.016; Fig. 8).

G-CSF significantly increases Cx43 expression in CHF-rabbit hearts. a Respresentative immunofluorescence detection of Cx43 (bright red fluorescing structures) in a frozen rabbit LV section (failing, G-CSF treated). Magnification ×200. b Mean relative Cx43 positive areas (related to cardiomyocyte areas; i.e., areas of red autofluorescence in a) in LV sections from sham and CHF hearts treated with G-CSF or left untreated, respectively, as indicated (n numbers of animals investigated)
Discussion
The main and novel finding of the present study is that chronic G-CSF therapy leads to a reduction of repolarization reserve in a non-ischemic heart failure model. In animal models of ischemic heart failure, G-CSF has been demonstrated to cause beneficial effects on cardiac structure and pump function [11, 30, 41]. Our group previously showed that G-CSF therapy led to a higher density of arterial vessels in the infarct border zone as well as to an increased cardiac output in an experimental mouse model of myocardial infarction. Furthermore, a reduction of ventricular tachycardia potentially related to an alleviated downregulation of connexin 43 expression in the infarct border zone was found. In agreement with these findings, G-CSF had similar effects on arterial and capillary density and showed a clear trend to slower progression of echocardiographically detected LV dysfunction in the non-ischemic rabbit model of heart failure.
However, in contrast to the potentially beneficial effect on ventricular tachycardia due to macro-reentry in the post-myocardial infarction model [30], the present study revealed a reduced repolarization reserve increasing proarrhythmia in pacing-induced heart failure. Chronic G-CSF therapy led to a prolongation of action potential duration and an increase of spatial and temporal dispersion of repolarization, thus augmenting the spontaneous occurrence of polymorphic ventricular tachyarrhythmias due to triggered activity despite the increased expression of connexin 43.
Repolarization reserve
The development of proarrhythmia is an individual response to a repolarization prolonging drug depending on the so-called “repolarization reserve” [44]. This concept describes that known and yet unknown, e.g., genetic defects or other modifying factors set the stage for an abnormal response to a repolarization prolonging drug. When the repolarization process is influenced by clinical risk factors such as heart failure [37], superimposition of an I Kr-blocking drug may produce marked action potential prolongation, resulting in triggered activity [45]. Erythromycin-induced proarrhythmia is an important clinical problem. The FDA has published (http://www.fda.org) several reports of erythromycin-related proarrhythmia. Our data (MAP90, overall dispersion, temporal dispersion and incidence of ventricular tachyarrhythmias) clearly demonstrate that administration of an I Kr-blocking drug led to a greater effect on repolarization in failing as compared with normal hearts, thus emphasizing the proarrhythmic role of heart failure as a contributing risk factor for a reduced repolarization reserve. Whereas the underlying mechanisms of pacing-induced heart failure and possible electrophysiologic changes are largely elusive [18], our data clearly show that CHF acts as an amplifier that increases the probability of proarrhythmia [44]. G-CSF further reduced repolarization reserve in the present model. This led to a significant increase of ventricular tachyarrhythmias and may indicate the potential risk of combining G-CSF with repolarization prolonging drugs in non-ischemic cardiomyopathy since these risk factors act synergistic on myocardial repolarization reserve.
Predictors of proarrhythmia
Prolongation of the action potential duration in failing hearts has been reported before in various experimental models [14, 59] as well as in humans [33]. Although it has previously been shown, that the extent of QT prolongation does not necessarily correlate with the degree of proarrhythmia [39], prolongation of repolarization acts as an important precondition for the generation of EAD and may be associated with an increased dispersion of repolarization. Thus, to some degree, action potential prolongation in CHF may be regarded as a form of acquired LQT syndrome. This is underlined by the occurrence of polymorphic ventricular tachyarrhythmias resembling torsade de pointes in the present study and previous studies [14, 37] and in patients suffering from heart failure [25]. In the presence of drug-related I Kr blockade, hearts that were chronically treated with G-CSF showed a further increase in action potential duration.
It has previously been demonstrated that erythromycin increases dispersion of repolarization [2], which has been accepted as an important factor for the initiation of polymorphic ventricular tachyarrhythmias [61]. We were able to show a more marked increase of spatial and temporal dispersion of repolarization in heart failure as compared to control. In normal and failing hearts, G-CSF treatment further increased dispersion, again pointing to a proarrhythmic effect of G-CSF. An increased QT dispersion in patients with heart failure has been linked to an increased risk of sudden cardiac death [4]. In addition, Tomaselli et al. [58] found a larger regional dispersion of refractoriness in patients with cardiomyopathy, which was comparable to that measured in patients with long QT syndrome. Both failing and control hearts chronically pre-treated with G-CSF showed an excessively increased temporal dispersion after acute application of the I Kr-blocking agent erythromycin in our study. This beat-to-beat variability may be a useful predictor for the occurrence of proarrhythmia [24] since a direct correlation between an elevated temporal dispersion and the occurrence of proarrhythmia has been demonstrated before [38, 57]. In our study, the marked increase of temporal dispersion, coinciding with action potential prolongation and an increase of spatial dispersion, was associated with an elevated incidence of EAD and VT in G-CSF hearts.
G-CSF effects on arrhythmogenesis and vascularization
Recent studies revealed a protective and/or regenerative effect of G-CSF treatment in the setting of acute myocardial infarction [15, 30, 56]. Adverse remodeling and, in the case of our previous work [30], ventricular tachyarrhythmias could be suppressed by application of G-CSF in a murine model of myocardial infarction. However, arrhythmia in this setting is due to re-entrant mechanisms in the border zone of infarction caused by slowing of conduction which may be due to a reduction of connexin 43 expression in this area [42]. Together with work by van Rijen et al. [60] who demonstrated that a reduction of connexin 43 expression in hearts of transgenic mice of 50% is not sufficient to cause inducibility of ventricular tachyarrhythmias, whereas an extent of 85% reduction or more is clearly proarrhythmic, our data [30] suggest that the antiarrhythmic effect of G-CSF was due to an alleviation of the reduced connexin 43 expression in the infarct border zone. In the present investigation, connexin 43 expression was found to be reduced by only 45% in failing rabbit hearts and G-CSF abrogated this effect in agreement with our earlier findings and those of others [31]. However, apart from different mechanisms causing arrhythmias (reentry vs. triggered activity) we assume that the extent of alterations in connexin 43 expression in failing G-CSF pre-treated hearts was not sufficient enough to compensate G-CSF’s negative influence on repolarization reserve in the present model. The discrepancy between the data presented here and those published earlier [30] might be also due to the fact that G-CSF was applied transiently (for 6 days) around the time of infarction in the case of the mouse study while it was applied chronically in a higher dosage until the killing of the animals in the present work. However, with respect to the apparent slowdown of the progression of heart failure in the G-CSF-treated animals the increase of connexin 43 expression might have been a contributing factor as it has been shown for murine ischemic heart failure that a decrease of the expression of connexin 43 by 50% already compromises the protective effect of ischemic preconditioning [54] which, again, in the porcine heart is based on the prevention of connexin 43 dephosphorylation during subsequent sustained ischemia [53].
Regarding the mechanism of G-CSF-mediated proarrhythmia in the present study, increased numbers of neutrophils, liberating increased amounts of platelet activating factor, might play a role as suggested by findings in a canine model, where activation of neutrophils was arrhythmogenic [22]. Of note, platelet activating factor (PAF), liberated by neutrophils caused an increase in action potential duration, and EAD in the same model [23]. This effect might result from a direct inhibiting effect of PAF on potassium channels [3]. However, data from our laboratory suggest that there are most likely no differences between the numbers of neutrophils in cardiac tissue samples of the four rabbit groups. In agreement with this observation neither pacing nor G-CSF treatment significantly increased levels of PAF in rabbit cardiac tissue as assessed by ELISA (data not shown).
Correspondingly, the proarrhythmic effect could also be related to a so far unknown direct effect of G-CSF on ion channels involved in the repolarization process, as G-CSF receptor protein is expressed by cardiomyocytes [15, 30].
Irrespective of the etiology of the disease, this work is, with respect to the increased arterial density caused by G-CSF in failing rabbit hearts, in agreement with our previous investigation of murine ischemic heart failure [30]. Similar effects on arterialization (e.g., [11]) as well as capillary density [26] in rodent models of ischemic heart failure were also observed by others. Regarding the positive effect of G-CSF on arterial density in the hearts of sham-operated rabbits corresponding data also demonstrating an increase of arterial/arteriolar density in normal hearts were published for other growth factors such as acidic fibroblast growth factor [13, 48] and vascular endothelial growth factor 121 [50] as well as for the thyroid hormone analog 3,5-diiodothyropropionic acid (DIPTA) [63]. Carrao and coworkers [8] determined an increased coronary blood flow in non-ischemic rat hearts after G-CSF treatment which, according to their assumption, reflects an increased collateral growth. They provided data suggesting a potential role of reactive oxygen (ROS) species, which are synthesized in cardiomyocytes upon G-CSF treatment in vitro apparently leading to the secretion of angiogenic factors. In combination with our data regarding increased expression of connexin 43 in G-CSF treated hearts (Fig. 8) this interpretation is in agreement with an investigation showing that reduced connexin 43 expression by cardiomyocytes is correlated with a deficit in ROS formation and a corresponding loss of cardioprotection [17]. However, whether this hypothetical mechanism also plays a role in the G-CSF effect in the rabbit pacing model is so far an open question.
Assuming that neutrophils and other bone marrow-derived cells are not significantly enriched in rabbit hearts, neither by pacing nor by G-CSF treatment (see above), a direct growth promoting effect of the cytokine on vessels is more likely than mechanisms based on paracrine effects or on differentiating bone marrow-derived vascular progenitor cells as it was excluded by our previous investigation for infarcted mouse hearts already [30]. This hypothesis fits with investigations which demonstrated stimulating effects of G-CSF on the proliferation and migration of endothelial cells [6] and vascular smooth muscle cells [9, 10]. In agreement, the recruitment of the latter by capillaries has been discussed as a potential mechanism contributing to the formation of collateral arteries (for review: [7, 51]. Furthermore, a pro-survival effect might play a role as STAT3 [15] as well as PI3K/Akt signaling [29] which both are stimulated by G-CSF (for review: [28] mediate a pro-survival effect of the cytokine on endothelial cells [15]. In agreement with this interpretation, the knockdown of STAT3 signaling causes a gradual decrease of capillaries even in normal adult hearts [21].
Conclusion
In the non-ischemic pacing-induced rabbit CHF model, chronic G-CSF treatment led to a significant reduction of repolarization reserve demonstrated by an increase in spatial and temporal dispersion of repolarization facilitating the occurrence of ventricular tachyarrhythmias due to triggered activity. This action is despite moderately beneficial effects on connexin 43 expression. This increase in arrhythmogenesis might limit its suitability as a therapeutic agent in the setting of heart failure. Thus, therapeutic approaches with cytokines in heart failure should be carefully monitored with regard to myocardial repolarization. In addition, G-CSF showed moderate beneficial effects on various parameters potentially related to myocardial contractility such as arteriogenesis, angiogenesis and connexin 43 expression in failing hearts.
References
Amankwa K, Krishnan SC, Tisdale JE (2004) Torsades de pointes associated with fluoroquinolones: importance of concomitant risk factors. Clin Pharmacol Ther 75:242–247. doi:10.1016/j.clpt.2003.11.376
Antzelevitch C, Sun ZQ, Zhang ZQ, Yan GX (1996) Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J Am Coll Cardiol 28:1836–1848. doi:10.1016/S0735-1097(96)00377-4
Barbuti A, Ishii S, Shimizu T, Robinson RB, Feinmark SJ (2002) Block of the background K(+) channel TASK-1 contributes to arrhythmogenic effects of platelet-activating factor. Am J Physiol Heart Circ Physiol 282:H2024–H2030. doi:10.1152/ajpheart.00956.2001
Bonnar CE, Davie AP, Caruana L, Fenn L, Ogston SA, McMurray JJ, Struthers AD (1999) QT dispersion in patients with chronic heart failure: beta blockers are associated with a reduction in QT dispersion. Heart 81:297–302
Brennan M, Palaniswami M, Kamen P (2002) Poincare plot interpretation using a physiological model of HRV based on a network of oscillators. Am J Physiol Heart Circ Physiol 283:H1873–H1886. doi:10.1152/ajpheart.00405.2000
Bussolino F, Ziche M, Wang JM, Alessi D, Morbidelli L, Cremona O, Bosia A, Marchisio PC, Mantovani A (1991) In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J Clin Invest 87:986–995. doi:10.1172/JCI115107
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395. doi:10.1038/74651
Carrao AC, Chilian WM, Yun J, Kolz C, Rocic P, Lehmann K, van den Wijngaard JP, van Horssen P, Spaan JA, Ohanyan V, Pung YF, Buschmann I (2009) Stimulation of coronary collateral growth by granulocyte stimulating factor: role of reactive oxygen species. Arterioscler Thromb Vasc Biol 29:1817–1822. doi:10.1161/ATVBAHA.109.186445
Chen X, Kelemen SE, Autieri MV (2004) AIF-1 expression modulates proliferation of human vascular smooth muscle cells by autocrine expression of G-CSF. Arterioscler Thromb Vasc Biol 24:1217–1222. doi:10.1161/01.ATV.0000130024.50058.de
Chen X, Kelemen SE, Autieri MV (2005) Expression of granulocyte colony-stimulating factor is induced in injured rat carotid arteries and mediates vascular smooth muscle cell migration. Am J Physiol Cell Physiol 288:C81–C88. doi:10.1152/ajpcell.00322.2004
Deindl E, Zaruba MM, Brunner S, Huber B, Mehl U, Assmann G, Hoefer IE, Mueller-Hoecker J, Franz WM (2006) G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J 20:956–958. doi:10.1096/fj.05-4763fje
Eckardt L, Haverkamp W, Borggrefe M, Breithardt G (1998) Experimental models of torsade de pointes. Cardiovasc Res 39:178–193 (pii:S0008-6363(98)00043-1)
Fernandez B, Buehler A, Wolfram S, Kostin S, Espanion G, Franz WM, Niemann H, Doevendans PA, Schaper W, Zimmermann R (2000) Transgenic myocardial overexpression of fibroblast growth factor-1 increases coronary artery density and branching. Circ Res 87:207–213
Frommeyer G, Milberg P, Witte P, Stypmann J, Koopmann M, Lucke M, Osada N, Breithardt G, Fehr M, Eckardt L (2011) A new mechanism preventing proarrhythmia in chronic heart failure: rapid phase-III repolarization explains the low proarrhythmic potential of amiodarone in contrast to sotalol in a model of pacing-induced heart failure. Eur J Heart Fail 13:1060–1069. doi:10.1093/eurjhf/hfr107
Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, Ohtsuka M, Matsuura K, Sano M, Nishi J, Iwanaga K, Akazawa H, Kunieda T, Zhu W, Hasegawa H, Kunisada K, Nagai T, Nakaya H, Yamauchi-Takihara K, Komuro I (2005) G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med 11:305–311. doi:10.1038/nm1199
Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, Escande D, Franz M, Malik M, Moss A, Shah R (2000) The potential for QT prolongation and proarrhythmia by non-antiarrhythmic drugs: clinical and regulatory implications. Report on a policy conference of the European Society of Cardiology. Eur Heart J 21:1216–1231. doi:10.1053/euhj.2000.2249
Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di LF, Schulz R, Heusch G (2005) Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 97:583–586. doi:10.1161/01.RES.0000181171.65293.65
Heusch G (2011) Heart rate and heart failure. Not a simple relationship. Circ J 75:229–236 (pii:JSTJSTAGE/circj/CJ-10-0925)
Heusch P, Aker S, Boengler K, Deindl E, van de Sand A, Klein K, Rassaf T, Konietzka I, Sewell A, Menazza S, Canton M, Heusch G, Di LF, Schulz R (2010) Increased inducible nitric oxide synthase and arginase II expression in heart failure: no net nitrite/nitrate production and protein S-nitrosylation. Am J Physiol Heart Circ Physiol 299:H446–H453. doi:10.1152/ajpheart.01034.2009
Heusch P, Canton M, Aker S, van de Sand A, Konietzka I, Rassaf T, Menazza S, Brodde OE, Di LF, Heusch G, Schulz R (2010) The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits. Br J Pharmacol 160:1408–1416. doi:10.1111/j.1476-5381.2010.00793.x
Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H (2004) Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95:187–195. doi:10.1161/01.RES.0000134921.50377.61
Hoffman BF, Feinmark SJ, Guo SD (1997) Electrophysiologic effects of interactions between activated canine neutrophils and cardiac myocytes. J Cardiovasc Electrophysiol 8:679–687
Hoffman BF, Guo SD, Feinmark SJ (1996) Arrhythmias caused by platelet activating factor. J Cardiovasc Electrophysiol 7:120–133
Hondeghem LM, Carlsson L, Duker G (2001) Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 103:2004–2013
Hondeghem LM, Snyders DJ (1990) Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 81:686–690
Imazuru T, Matsushita S, Hyodo K, Tokunaga C, Kanemoto S, Enomoto Y, Watanabe Y, Hiramatsu Y, Sakakibara Y (2009) Erythropoietin enhances arterioles more significantly than it does capillaries in an infarcted rat heart model. Int Heart J 50:801–810 (pii:JST.JSTAGE/ihj/50.801)
Jia N, Dong P, Huang Q, Jin W, Zhang J, Dai Q, Liu S (2009) Cardioprotective effects of granulocyte colony-stimulating factor in angiotensin II-induced cardiac remodelling. Clin Exp Pharmacol Physiol 36:262–266. doi:10.1111/j.1440-1681.2008.05052.x
Klocke R, Kuhlmann MT, Scobioala S, Schabitz WR, Nikol S (2008) Granulocyte colony-stimulating factor (G-CSF) for cardio- and cerebrovascular regenerative applications. Curr Med Chem 15:968–977
Kojima H, Otani A, Oishi A, Makiyama Y, Nakagawa S, Yoshimura N (2011) Granulocyte colony-stimulating factor attenuates oxidative stress-induced apoptosis in vascular endothelial cells and exhibits functional and morphologic protective effect in oxygen-induced retinopathy. Blood 117:1091–1100. doi:10.1182/blood-2010-05-286963
Kuhlmann MT, Kirchhof P, Klocke R, Hasib L, Stypmann J, Fabritz L, Stelljes M, Tian W, Zwiener M, Mueller M, Kienast J, Breithardt G, Nikol S (2006) G-CSF/SCF reduces inducible arrhythmias in the infarcted heart potentially via increased connexin43 expression and arteriogenesis. J Exp Med 203:87–97. doi:10.1084/jem.20051151
Kuwabara M, Kakinuma Y, Katare RG, Ando M, Yamasaki F, Doi Y, Sato T (2007) Granulocyte colony-stimulating factor activates Wnt signal to sustain gap junction function through recruitment of beta-catenin and cadherin. FEBS Lett 581:4821–4830. doi:10.1016/j.febslet.2007.09.007
Lassaletta AD, Chu LM, Sellke FW (2011) Therapeutic neovascularization for coronary disease: current state and future prospects. Basic Res Cardiol. doi:10.1007/s00395-011-0200-1
Lehmann MH, Hardy S, Archibald D, quart B, MacNeil DJ (1996) Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation 94:2535–2541
Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Ogino A, Maruyama R, Nakagawa M, Minatoguchi S, Fujiwara T, Fujiwara H (2007) Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest 87:440–455. doi:10.1038/labinvest.3700530
Milberg P, Fink M, Pott C, Frommeyer G, Biertz J, Osada N, Stypmann J, Mönnig G, Koopmann M, Breithardt G, Eckardt L (2011) ICa block leads to suppression of early afterdepolarizations and reduction of transmural dispersion of repolarization in a whole heart model of chronic heart failure. Br J Pharmacol. doi:10.1111/j.1476-5381.2011.01721
Milberg P, Eckardt L, Bruns HJ, Biertz J, Ramtin S, Reinsch N, Fleischer D, Kirchhof P, Fabritz L, Breithardt G, Haverkamp W (2002) Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 303:218–225. doi:10.1124/jpet.102.037911
Milberg P, Fleischer D, Stypmann J, Osada N, Monnig G, Engelen MA, Bruch C, Breithardt G, Haverkamp W, Eckardt L (2007) Reduced repolarization reserve due to anthracycline therapy facilitates torsade de pointes induced by IKr blockers. Basic Res Cardiol 102:42–51. doi:10.1007/s00395-006-0609-0
Milberg P, Frommeyer G, Kleideiter A, Fischer A, Osada N, Breithardt G, Fehr M, Eckardt L (2011) Antiarrhythmic effects of free polyunsaturated fatty acids in an experimental model of LQT2 and LQT3 due to suppression of early afterdepolarizations and reduction of spatial and temporal dispersion of repolarization. Heart Rhythm. doi:10.1016/j.hrthm.2011.03.058
Milberg P, Ramtin S, Monnig G, Osada N, Wasmer K, Breithardt G, Haverkamp W, Eckardt L (2004) Comparison of the in vitro electrophysiologic and proarrhythmic effects of amiodarone and sotalol in a rabbit model of acute atrioventricular block. J Cardiovasc Pharmacol 44:278–286 (pii:00005344-200409000-00002)
Milting H, Jacob M, Kassner A, Heimann P, Mannherz HG, Becker G, Meyer HE, Bothig D, Arusoglu L, Morshuis M, Korfer R, El BA (2004) The structural examination of myocardial samples from patients with end-stage heart failure supported by ventricular assist devices using electron microscopy and amino acid analysis reveals low degree of reverse remodeling. J Heart Lung Transplant 23:396–404. doi:10.1016/S1053-2498(03)00205-5
Okada H, Takemura G, Li Y, Ohno T, Li L, Maruyama R, Esaki M, Miyata S, Kanamori H, Ogino A, Nakagawa M, Minatoguchi S, Fujiwara T, Fujiwara H (2008) Effect of a long-term treatment with a low-dose granulocyte colony-stimulating factor on post-infarction process in the heart. J Cell Mol Med 12:1272–1283. doi:10.1111/j.1582-4934.2008.00294.x
Peters NS, Coromilas J, Severs NJ, Wit AL (1997) Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 95:988–996
Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM (2004) Oral erythromycin and the risk of sudden death from cardiac causes. N Engl J Med 351:1089–1096. doi:10.1056/NEJMoa040582
Roden DM (1998) Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol 21:1029–1034
Roden DM (2006) Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med 259:59–69. doi:10.1111/j.1365-2796.2005.01589.x
Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y (2008) Heart disease and stroke statistics-2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 117:e25–e146. doi:10.1161/CIRCULATIONAHA.107.187998
Rubart M, Pressler ML, Pride HP, Zipes DP (1993) Electrophysiological mechanisms in a canine model of erythromycin-associated long QT syndrome. Circulation 88:1832–1844
Safi J Jr, DiPaula AF Jr, Riccioni T, Kajstura J, Ambrosio G, Becker LC, Anversa P, Capogrossi MC (1999) Adenovirus-mediated acidic fibroblast growth factor gene transfer induces angiogenesis in the nonischemic rabbit heart. Microvasc Res 58:238–249. doi:10.1006/mvre.1999.2165
Sanganalmath SK, Abdel-Latif A, Bolli R, Xuan YT, Dawn B (2011) Hematopoietic cytokines for cardiac repair: mobilization of bone marrow cells and beyond. Basic Res Cardiol 106:709–733. doi:10.1007/s00395-011-0183-y
Schalch P, Rahman GF, Patejunas G, Goldschmidt RA, Carbray J, Retuerto MA, Kim D, Esser K, Crystal RG, Rosengart TK (2004) Adenoviral-mediated transfer of vascular endothelial growth factor 121 cDNA enhances myocardial perfusion and exercise performance in the nonischemic state. J Thorac Cardiovasc Surg 127:535–540. doi:10.1016/j.jtcvs.2003.06.015
Schaper W (2009) Collateral circulation: past and present. Basic Res Cardiol 104:5–21. doi:10.1007/s00395-008-0760-x
Schulz R, Aker S, Belosjorow S, Konietzka I, Rauen U, Heusch G (2003) Stress kinase phosphorylation is increased in pacing-induced heart failure in rabbits. Am J Physiol Heart Circ Physiol 285:H2084–H2090. doi:10.1152/ajpheart.01038.2002
Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G (2003) Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J 17:1355–1357. doi:10.1096/fj.02-0975fje
Schwanke U, Konietzka I, Duschin A, Li X, Schulz R, Heusch G (2002) No ischemic preconditioning in heterozygous connexin43-deficient mice. Am J Physiol Heart Circ Physiol 283:H1740–H1742. doi:10.1152/ajpheart.00442.2002
Scobioala S, Klocke R, Kuhlmann M, Tian W, Hasib L, Milting H, Koenig S, Stelljes M, El-Banayosy A, Tenderich G, Michel G, Breithardt G, Nikol S (2008) Up-regulation of nestin in the infarcted myocardium potentially indicates differentiation of resident cardiac stem cells into various lineages including cardiomyocytes. FASEB J 22:1021–1031. doi:10.1096/fj.07-8252com
Sugano Y, Anzai T, Yoshikawa T, Maekawa Y, Kohno T, Mahara K, Naito K, Ogawa S (2005) Granulocyte colony-stimulating factor attenuates early ventricular expansion after experimental myocardial infarction. Cardiovasc Res 65:446–456. doi:10.1016/j.cardiores.2004.10.008
Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de PG, van OJ, Volders PG, Vos MA (2004) Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation 110:2453–2459. doi:10.1161/01.CIR.0000145162.64183.C8
Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E (1994) Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 90:2534–2539
Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, Kodama I (2000) Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res 48:300–309 (pii:S0008-6363(00)00180-2)
van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM (2004) Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 109:1048–1055. doi:10.1161/01.CIR.0000117402.70689.75
Verduyn SC, Vos MA, van der Zande J, van Der Hulst FF, Wellens HJ (1997) Role of interventricular dispersion of repolarization in acquired torsade-de-pointes arrhythmias: reversal by magnesium. Cardiovasc Res 34:453–463 (pii:S0008-6363(97)00067-9)
Volders PG, Stengl M, van Opstal JM, Gerlach U, Spatjens RL, Beekman JD, Sipido KR, Vos MA (2003) Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation 107:2753–2760. doi:10.1161/01.CIR.0000068344.54010.B3
Wang X, Zheng W, Christensen LP, Tomanek RJ (2003) DITPA stimulates bFGF, VEGF, angiopoietin, and Tie-2 and facilitates coronary arteriolar growth. Am J Physiol Heart Circ Physiol 284:H613–H618. doi:10.1152/ajpheart.00449.2002
Yang T, Roden DM (1996) Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation 93:407–411
Acknowledgments
We thank Dr. Tilmann Spieker, Gerhard-Domagk-Institute for Pathology, Westfälische Wilhelms-University Münster, for worthwhile scientific discussions and Sezen Maleki and Birgit Jaxy for excellent technical assistance. This study was supported by the Dr. Peter Osypka Foundation; Lörrach, Germany. Lars Eckardt holds the Peter Osypka Professorship of Experimental and Clinical Electrophysiology at the University of Münster, Münster, Germany. Further support was provided by the Hans und Gertie Fischer-Stiftung (Essen, Germany) to Sigrid Nikol and Rainer Klocke.
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Milberg and R. Klocke contributed equally.
Rights and permissions
About this article
Cite this article
Milberg, P., Klocke, R., Frommeyer, G. et al. G-CSF therapy reduces myocardial repolarization reserve in the presence of increased arteriogenesis, angiogenesis and connexin 43 expression in an experimental model of pacing-induced heart failure. Basic Res Cardiol 106, 995–1008 (2011). https://doi.org/10.1007/s00395-011-0230-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-011-0230-8