
Abstract
Background
Non-vitamin K antagonist oral anticoagulants (NOACs) are at least as effective and safe as vitamin K antagonists (VKAs) for stroke prevention in atrial fibrillation (AF). All pivotal trials have compared NOACs to warfarin. However, other VKAs are commonly used, for instance phenprocoumon.
Patients and methods
A retrospective cohort study using a German claims database assessed the comparative risks of bleeding leading to hospitalization during therapy with NOACs and phenprocoumon in AF patients. Endpoints consisted of major bleeding, gastrointestinal bleeding, and any bleeding. Data were collected from January 1, 2013 to March 31, 2015. Patients newly initiated on dabigatran, apixaban, rivaroxaban, or phenprocoumon were included. Hazard Ratios for bleeding events were derived from Cox proportional hazard models, adjusting for differences in baseline characteristics. Propensity score matching was performed as a sensitivity analysis.
Results
A total of 35,013 patients were identified, including 3138 on dabigatran, 3633 on apixaban, 12,063 on rivaroxaban, and 16,179 on phenprocoumon. Patients prescribed apixaban or phenprocoumon were older compared to those on dabigatran or rivaroxaban and had a higher CHA2DS2-VASc score. After adjusting for baseline confounders, apixaban was associated with lower risks of major bleeding (HR 0.68, 95% CI 0.51–0.90, p = 0.008), gastrointestinal bleeding (HR 0.53, 95% CI 0.39–0.72, p < 0.001), and any bleeding (HR 0.80, 95% CI 0.70–0.92, p = 0.002) compared to phenprocoumon. There were no significant differences in bleeding risk between dabigatran and phenprocoumon. Rivaroxaban was associated with more gastrointestinal bleeding (HR 1.39, 95% CI 1.21–1.60, p < 0.001) and any bleeding (HR 1.19, 95% CI 1.10–1.28, p < 0.001). Sensitivity analysis using propensity score matching confirmed these observations.
Conclusions
Apixaban therapy is associated with a significantly reduced risk of bleeding compared to phenprocoumon. Bleeding risk with dabigatran was similar to that of phenprocoumon but bleeding risk with rivaroxaban was higher.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in clinical practice with an overall prevalence of around 2% in the general population [1]. AF is associated with significant morbidity and mortality, mostly due to thromboembolic complications such as stroke or peripheral embolism and heart failure. The risk of stroke can be effectively reduced by oral anticoagulation (OAC) which in the past was predominantly achieved by administering vitamin K antagonists (VKAs). Since 2009, four non-vitamin K oral antagonists (NOACs) have been tested and approved for stroke prevention in AF. These large randomized controlled trials have demonstrated that NOAC therapy is at least as effective as and probably safer than treatment with VKAs [2–5]. All pivotal trials have evaluated NOACs against therapy with warfarin. However, in some regions of the world, the most commonly used VKA is phenprocoumon, for instance in Germany. This VKA differs in pharmacokinetic and pharmacodynamic properties from warfarin; most notably, phenprocoumon has a very long elimination half-life (110–130 h) compared to warfarin (35–40 h) [6]. Although German authorities considered phenprocoumon to be equivalent to warfarin with respect to the observed anticoagulant effects in the phase III trials, no actual data exist comparing this specific VKA to any of the NOACs. Hence, the current real-world study aimed to assess and compare bleeding profiles among German patients with non-valvular AF who were new users of phenprocoumon or apixaban, dabigatran, or rivaroxaban.
Methods
Study design and data source
The retrospective CARBOS study [CompArative Risk of major Bleeding with new Oral anticoagulantS (NOACs) and phenprocoumon in patients with atrial fibrillation] is based on an anonymized research database from the Health Risk Institute (HRI) [7, 8]. As a post-authorization safety study (PASS), the study is registered at the European Medicines Agency and the protocol is published under http://www.encepp.eu/encepp/viewResource.htm?id=14218. The HRI performs independent statistical analyses on anonymized claims data for patient-level risk prediction, outcome research, and patient safety. The HRI database comprises longitudinal information on medical and drug claims from an age- and gender-representative sample of about 4 million statutory health-insured subjects in Germany, representing approximately 5% of the total population.
Data available from each medical claim include date/quarter of service, place of service, diagnoses [International Statistical Classification of Diseases and Related Health Problems, 10th revision, German Modification (ICD-10-GM)], and procedures performed/services rendered. Data available for each drug claim include the agent dispensed [as set forth by the Anatomical Therapeutic Chemical (ATC) System], dispensing/prescription date, and quantity dispensed. Selected demographic and eligibility information (including age/year of birth, sex, dates of enrollment) is also available for subjects in the HRI Database. All data can be arrayed to provide a detailed chronology of medical and pharmacy series used by each insured member over time.
All patient-level data in the HRI Research Database are de-identified to comply with German data protection regulations. Use of the study database for health services research is therefore fully compliant with German federal law and, accordingly, IRB/ethical approval was not applicable.
Study population
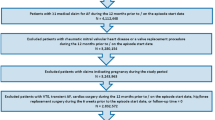
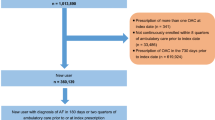
Adult patients (≥18 years) with non-valvular AF were identified who were new users of apixaban, dabigatran, rivaroxaban, and phenprocoumon between January 1, 2013 and December 31, 2014 (Fig. 1). A new user was required to have no prior prescription for any of the above-listed substances in the 12 months before initiation of medication. If a patient ever used NOACs or phenprocoumon during the study period, the first prescription was defined as the index medication and the date of this first prescription as the index date. At the time of data collection, edoxaban was not yet approved for stroke prevention in AF, and hence no data for this anticoagulant were available. Patients were excluded if they were not continuously represented in the HRI Database for at least 1 year prior to January 1, 2013, which was defined as the baseline period. All patients were required to have at least one primary or secondary hospital discharge diagnosis of AF in the previous or same quarter of the index date or—alternatively—at least two ambulatory verified diagnoses of AF in the period between January 1, 2010 and the index date. Patients with valvular AF, deep vein thrombosis, hemodialysis, pregnancy, or anticoagulation therapy (i.e., heparin, low-molecular weight heparin, vitamin K antagonists, or NOACs) for any other indication during the four quarters prior to or on the index date were excluded.

Patient selection flowchart
Study endpoints
The primary study endpoint was a documented major bleeding event. Secondary endpoints were gastrointestinal bleeding events or any bleeding events. A composite net clinical outcome consisting of ischemic stroke, systemic embolism, or major bleeding was defined as a tertiary endpoint. Bleeding events that occurred on treatment, defined as the time from the first prescription until the end of the study period, discontinuation of treatment, death, end of continuous enrollment, or switching to another OAC, were included.
Major bleeding consisted of an emergency hospital admission with an ICD-10-GM hospital discharge diagnosis. Gastrointestinal bleeding was defined as bleeding at any time during exposure time with localization in the gastrointestinal tract and documented ICD-10-GM hospital discharge diagnosis. Any bleeding was defined using pre-specified primary or secondary ICD-10-GM hospital discharge diagnoses at any time (see Table S1 of the Supplementary Appendix).
We used the drug claims to determine patient’s treatment periods, defined as the time from the initial prescription to the date with no residual days of drug supply. A maximum gap of 30 days between treatment periods was allowed. Patients were considered to be continuing on treatment as long as they had another medication prescription within 30 days of the end of the last treatment period. In addition to this conservative approach, we conducted a sensitivity analysis by changing the allowable gap to 2 days for NOACs and 10 days for phenprocoumon, with largely unchanged findings.
Statistical analysis
Baseline characteristics were presented descriptively. Unadjusted event rates were estimated for each treatment group and were expressed per 100 person-years. Cox proportional hazard models were used to estimate the hazard ratios (HRs) of major bleeding, gastrointestinal bleeding, any bleeding, and net clinical outcome adjusted for pre-specified baseline demographics and clinical factors. The variables that entered the final models were chosen on medical considerations and by using Akaike Information Criteria feature selection process [9], i.e., some variables like age, sex, Charlson comorbidity index, number of hospitalizations, and HAS-BLED score were forced into the model, whereas the others were chosen on empirical basis. The proportional hazard assumption was tested on the basis of Schoenfeld residuals [10] and was valid for all outcomes.
Sensitivity analyses
First, to assess the impact of different dosages on the primary findings the risk of major bleeding, gastrointestinal bleeding, and any bleeding was compared to phenprocoumon only for those patients who received the highest approved dose of NOACs only (2 × 5 mg/day for apixaban, 2 × 150 mg/day for dabigatran, 1 × 20 mg/day for rivaroxaban). Second, the respective risks of different bleeding events for each treatment were compared when prescribed in the study period or until death or the end of the insurance status. Hence, the date of a switch or of discontinuation of the OAC treatment was not used as a censoring date. Instead, the exposure times of patients who switched from one substance to another were assessed based on their actual exposure time under each successive anticoagulant they received during follow-up. Additionally, we applied a marginal structural model of Cox proportional hazards with inverse probability treatment weighting (MSM Cox PH) [11, 12]. This model allows to obtain unbiased estimates of treatment effects on outcome variables, when (i) the treatment changes over time and (ii) in the presence of time-dependent covariates that may simultaneously be confounders (possibly affected by prior treatment) and intermediate variables (predict both subsequent treatment and subsequent outcome). This analysis was performed to assess whether primary findings using the on-treatment approach would be affected by a different censoring process. Third, propensity score matching was used as an alternative to adjustment for baseline characteristics by means of a regression model [13]. Three matched cohorts (apixaban vs. phenprocoumon, dabigatran vs. phenprocoumon, and rivaroxaban vs. phenprocoumon) were created using 1:1 propensity score matching without replacement and with a caliper of 0.01. Propensity scores for NOAC treatment were estimated using logistic regression which included information on the same baseline characteristics that were used in the main analysis. Standardized mean differences were used to assess the balance of baseline characteristics after matching. A standardized difference <10% indicates a negligible difference in baseline characteristics and balanced matched cohorts. A Cox proportional hazard model was used to compare endpoints in each of the propensity score-matched cohorts. Because all baseline characteristics were balanced after propensity score matching, the Cox model included only treatment (a NOAC or phenprocoumon) as the independent variable.
All analyses were conducted using SAS 9.3 (SAS Institute Inc.) and R 3.1.0. Statistical significance was assumed with a two-sided p value <0.05.
Results
Patient population
Among 35,013 eligible patients, 3633 (10.4%) were initiated on apixaban, 3138 (9.0%) on dabigatran, 12,063 (34.5%) on rivaroxaban, and 16,179 (46.2%) on phenprocoumon (Table 1). Patients prescribed phenprocoumon or apixaban were older compared to those initiated on dabigatran or rivaroxaban, and had on average a higher CHA2DS2-VASc score and more comorbidities. Subjects treated with apixaban had the highest HAS-BLED score, received most frequently drugs known to increase bleeding risk (antiplatelets or NSAIDs), and most frequently had a history of ischemic stroke or TIA (Table 1). The mean follow-up for patients initiated on apixaban was 218 days, dabigatran 261 days, rivaroxaban 258 days, and phenprocoumon 280 days.
Safety outcomes
Figure 2 displays the unadjusted event rates, adjusted hazard ratios, and the corresponding forest plots for each pairwise medication comparison (apixaban, dabigatran, and rivaroxaban each vs. phenprocoumon) for major, gastrointestinal, and any bleedings. For apixaban and dabigatran, event rates per 100 person-years of all bleeding events were lower than that for phenprocoumon, while for rivaroxaban the event rates for all types of bleeding were higher compared to phenprocoumon. After adjusting for baseline confounders, apixaban was associated with lower risks of major bleeding (HR 0.68, 95% CI 0.51–0.90, p = 0.008), gastrointestinal bleeding (HR 0.53, 95% CI 0.39–0.72, p < 0.001), and any bleeding (HR 0.80, 95% CI 0.70–0.92, p = 0.002) compared to phenprocoumon. There were no significant differences in the risk of different types of bleeding between dabigatran and phenprocoumon users. Rivaroxaban was associated with a higher risk of gastrointestinal bleeding (HR 1.39, 95% CI 1.20–1.59, p < 0.001) and any bleeding (HR 1.19, 95% CI 1.10–1.28, p < 0.001), whereas there was no significant difference in the risk of major bleeding compared to phenprocoumon.

Unadjusted event rates (per 100 person-years) and adjusted hazard ratios with 95% confidence intervals (CIs) for each pairwise comparison (apixaban, dabigatran, and rivaroxaban each vs. phenprocoumon)
Net clinical combined outcome
Table 2 displays the unadjusted event rates per 100 person-years and adjusted hazard ratios for each pairwise medication comparison (apixaban, dabigatran, and rivaroxaban each vs. phenprocoumon) for the combined endpoint ischemic stroke, systemic embolism, and major bleeding. There were no significant differences between any of the NOACs and phenprocoumon for this net clinical combined outcome measure.
Sensitivity analyses
Figure 3 shows the results of the sensitivity analysis considering only patients treated with the highest approved dose of apixaban (n = 2231; 61%), dabigatran (n = 1496; 48%), and rivaroxaban (n = 8379; 69%). For apixaban, in patients receiving the highest approved dose of 2 × 5 mg/day, the results were consistent with the main analysis, i.e., superiority of apixaban vs. phenprocoumon for all types of bleedings studied. For the net clinical outcome endpoint, the findings confirmed the main analysis (HR 0.86, 95% CI 0.71–1.04, p = 0.121).

Sensitivity analysis based on patients treated with the highest approved dose of apixaban, dabigatran, and rivaroxaban
In contrast to the main analysis, dabigatran 2 × 150 mg/day dose users had a significantly lower risk of major bleeding and any bleeding as well as net clinical outcome compared with phenprocoumon users (HR 0.50, 95% CI 0.27–0.95, HR 0.70, 95% CI 0.54–0.90 and HR 0.59, 95% CI 0.38–0.93, respectively). The sensitivity analysis with rivaroxaban 1 × 20 mg/day dose users was consistent with the findings from the main analysis, revealing a higher risk of gastrointestinal as well as any bleeding with no statistically significant difference for major bleeding. Results for the net clinical outcome were consistent with the results from the main analysis (HR 1.01, 95% CI 0.92–1.12, p = 0.792).
The second sensitivity analysis, based on all treatments prescribed in the study period or until death or the end of the insurance status, revealed results for apixaban and dabigatran which were consistent with the main analysis (Fig. 4). Results for rivaroxaban patients remained the same for all types of bleeding studied; however, rivaroxaban users carried a significantly increased risk for net clinical combined outcome (HR 1.12, 95% CI 1.04–1.21, p = 0.002).

Sensitivity analysis based on all treatments that occurred between the index date and the end of the study period, death, or end of continuous enrollment
As a final sensitivity analysis, propensity score matching was used to adjust for possible differences in the baseline characteristics among treatment groups. We created three matched cohorts using 1:1 propensity score matching: phenprocoumon versus apixaban (n = 7262), phenprocoumon versus dabigatran (n = 6250), and phenprocoumon versus rivaroxaban (n = 22,550) (Table 3). Following propensity score matching, the baseline demographics and clinical factors, including the risk scores (CHA2DS2-VASc and HAS-BLED), were balanced with all standardized differences less than 10% between the matched cohorts (Table 3). Figure 5 displays the adjusted hazard ratios for each pairwise medication comparison for propensity score matching analysis. For apixaban patients, the findings remained consistent with the main analysis, i.e., there was a significantly lower risk for all safety endpoints and no significant difference with respect to the net clinical combined outcome compared to phenprocoumon patients. For patients using dabigatran, the results were also consistent with the results from the main analysis, the only exception being the endpoint of major bleeding. Patients on dabigatran had a significantly lower risk of major bleeding compared to users of phenprocoumon. With respect to the net clinical combined endpoint, there was no significant difference between users of dabigatran and phenprocoumon (HR 0.80, 95% CI 0.61–1.04, p = 0.095). For rivaroxaban patients, the results revealed a significantly higher risk for all bleeding types studied as well as for the net clinical combined outcome (HR 1.18, 95% CI 1.04–1.35, p = 0.013), in line with the result of the main analysis.

Sensitivity analysis based on propensity score matching
Discussion
Main findings
The present study is the first to compare the safety profile of NOACs to that of phenprocoumon in a real-world setting comprising more than 35,000 patients with non-valvular AF. Apixaban was associated with significantly lower risks of major bleeding, gastrointestinal bleeding, and any bleeding compared to phenprocoumon. There were no significant differences in the bleeding risks between dabigatran and phenprocoumon users, whereas rivaroxaban therapy was associated with increased risk of gastrointestinal bleeding as compared to phenprocoumon.
Risk of major bleeding with NOAC versus VKA therapy
In Europe, different coumarins are used including warfarin, acenocoumarol, and phenprocoumon. For instance, in Germany, the most commonly prescribed VKA for stroke prevention in AF is phenprocoumon. This VKA differs from warfarin in several pharmacokinetic, pharmacodynamic, and pharmacogenetic properties [6]. Most notably, this coumarin has the longest elimination half-life of 110–130 h [14] compared to warfarin with a half-life of 35–40 h [6]. Direct comparative data of phenprocoumon versus NOACs have not been published so far, and thus it is mandatory to acquire safety data from real-world data in patients treated with phenprocoumon or NOACs.
The comparison of safety aspects, i.e., major bleeding events, from the randomized controlled trials between the respective NOAC and VKA is limited by differences in the definition of major bleeding among the studies and imbalances in possibly unobservable baseline variables which cannot be adjusted for in observational studies. As adjudication of safety outcomes is usually not feasible in retrospective studies, the present study used bleeding requiring hospital admission as the definition of major bleeding which was identified by the respective ICD-10-GM codes. A similar strategy was used by the FDA in a ‘Protocol for Assessment of Dabigatran and Selected Safety Outcomes’ to extract bleeding events [15].
In the present large real-world patient population, apixaban carried a lower risk for major bleeding events than phenprocoumon after adjusting for potential baseline confounders by various statistical methods including propensity score matching. This risk reduction (HR 0.68, 95% CI 0.51–0.90, p = 0.008) was consistent with that observed in the pivotal phase III trial of apixaban versus warfarin (HR 0.69, 95% CI 0.60–0.80; p < 0.001) [4]. Our observations are also in agreement with the recently published real-world data comparing warfarin to apixaban [16, 17]. For instance, Yao and coworkers, using data from a large US insurance database, found that apixaban was associated with a significantly lower risk for major bleedings than warfarin (HR 0.45, 95% CI 0.34–0.59, p < 0.001) [17].
In patients receiving dabigatran, rivaroxaban, or phenprocoumon, major bleeding events occurred at similar incidences. These findings are again consistent with the observations made in the two respective phase III trials of these two NOACs, suggesting that in real-world conditions safety levels similar to what has been seen in the controlled clinical trials can be achieved. Given the similarity of the comparative safety data in the NOAC trials and this real-world comparison with phenprocoumon, it seems unlikely that the predominant use of phenprocoumon in Germany would be responsible for differences in the comparative safety. Therefore, there is no reason to assume that the results of the clinical trials of the NOAC cannot be extrapolated to daily practice when phenprocoumon is used as a predominant VKA.
Our sensitivity analysis, taking into account only patients treated with the highest approved NOAC doses, yielded results consistent with the main analysis. Therefore, it can be excluded that the observed real-world safety profile of the NOACs is an overestimation due to frequent use of reduced dosing regimens of the respective NOAC. This is especially pertinent to apixaban as the reduced dosing regimen of which is half of the standard dose (2 × 2.5 mg/day). In this respect, it is important to note that apixaban proved superior in safety to warfarin even though, on average, the patients treated with apixaban were older and had more comorbidities including renal failure and previous stroke/TIA and had higher HAS-BLED score than patients receiving dabigatran or rivaroxaban.
Risk of gastrointestinal bleeding with NOAC versus VKA therapy
Elderly patients with AF in particular are at risk for gastrointestinal bleeding. In the phase III trials of NOACs, dabigatran (2 × 150 mg/day) and rivaroxaban carried a higher risk of gastrointestinal bleeding than warfarin [2, 3]. Besides reduced dose of dabigatran (2 × 110 mg/day), apixaban was the only NOAC for which the risk for this unwanted effect did not exceed that of warfarin (HR 0.89, 95% CI 0.70–1.15; p = 0.37) in the randomized clinical trials [4]. Our findings are consistent with these observations from the randomized studies. The risk for gastrointestinal bleeding was significantly lower for apixaban than that for phenprocoumon (HR 0.53, 95% CI 0.39–0.72, p < 0.001). Dabigatran use was associated with a risk comparable to that of phenprocoumon, whereas the use of rivaroxaban carried a significantly higher gastrointestinal bleeding risk than VKA. All of our observations are in line with those of previously published real-world datasets [17–19]. The observed effects for the secondary endpoint any bleeding were also consistent with the findings from the randomized studies and previously published real-world datasets. It can therefore be concluded that the safety data observed in the controlled clinical trials of the NOACs can also be taken as a valid reference for real-world treatment conditions.
Net clinical outcome
The combined endpoint of ischemic stroke, systemic embolism, and major bleeding was chosen due to limited number of patients in the database and the short follow-up period. Thus, the number of ischemic stroke events was estimated to be too low to allow a valid comparison of effectiveness of the NOACs. Clarification of the effectiveness of the NOACs must therefore await analysis of larger patient samples with longer follow-up periods.
Limitations of the study
Our study is subject to a number of limitations which are inherent to any retrospective data analysis. Despite all attempts to adjust for important baseline confounders by applying various statistical methods including propensity score matching, residual bias cannot entirely be excluded. However, the large size of the patient sample and the consistency with previously published real-world studies and clinical trials indicates that our analyses yielded robust results. Another concern may be the potential for coding errors inherent to retrospective analysis of claims databases. However, one can expect that residual bias associated with coding errors may be similar for all exposure groups and thus should not meaningfully influence the assessment of our outcomes. The unavailability of INR measurements and laboratory data on renal function represents another inherent limitation of our study.
References
Wilke T, Groth A, Mueller S, Pfannkuche M, Verheyen F, Linder R, Maywald U, Bauersachs R, Breithardt G (2013) Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace 15:486–493
Connolly SJ, Ezekowitz MD, Yusuf S et al (2009) Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 361:1139–1151
Patel MR, Mahaffey KW, Garg J, Pan G et al (2011) Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 365:883–891
Granger CB, Alexander JH, McMurray JJ et al (2011) Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 365:981–992
Giugliano RP, Ruff CT, Braunwald E et al (2013) Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med 369:2093–2104
Verhoef TI, Redekop WK, Daly AK et al (2013) Pharmacogenetic-guided dosing coumarin anticoagulants: algorithms for warfarin, acenocoumarol and phenprocoumon. Br J Clin Pharmacol 77(4):626–641
Andersohn F, Walker J (2016) Characteristics and external validity of the German Health Risk Institute (HRI) database. Pharmacoepidemiol Drug Saf. doi:10.1002/pds.3895
Swart E, Gothe H, Geyer S, Jaunzeme J, Maier B, Grobe TG, Ihle P (2015) German Society for Social Medicine and Prevention; German Society for Epidemiology. Good Practice of Secondary Data Analysis (GPS): guidelines and recommendations. Gesundheitswesen 777:120–126
Hastie T, Tibshirani R, Friedman J (2009) The elements of statistical learning: data mining, inference and prediction, 2nd edn. Springer, Berlin
Schoenfeld D (1982) Partial residuals for the proportional hazards regression model. Biometrika 69:239–241
Robins JM, Hernán MA, Brumback B (2000) Marginal structural models and causal inference in epidemiology. Epidemiology 11:550–560
Hernán MA, Brumback B, Robins JM (2002) Estimating the causal effect of zidovudine on CD4 count with a marginal structural model for repeated measures. Stat Med 2:689–170913
Rosenbaum PR, Rubin DB (1983) The central role of the propensity score in observational studies for causal effects. Biometrika 70:41–55
Jahnchen E, Meinertz T, Gilfrich HJ, Groth U, Martini A (1976) The enantiomers of phenprocoumon: pharmacodynamic and pharmacokinetic studies. Clin Pharmacol Ther 20:342–349
Alan SG, Daniel S et al. (2015) Mini-sentinel medical product assessment: a protocol for assessment of dabigatran. https://www.sentinelsystem.org/sites/default/files/Drugs/Assessments/Mini-Sentinel_Protocol-for-Assessment-of-Dabigatran_0.pdf
Lip GY, Keshishian A, Kamble S, Pan X, Mardekian J, Horblyuk R, Hamilton M (2016) Real-world comparison of major bleeding risk among non-valvular atrial fibrillation patients initiated on apixaban, dabigatran, rivaroxaban, or warfarin. A propensity score matched analysis. Thromb Haemost 116:975–986
Yao X, Abraham NS, Sangaralingham LR, Bellolio MF, McBane RD, Shah ND, Noseworthy PA (2016) Effectiveness and safety of dabigatran, rivaroxaban, and apixaban versus warfarin in nonvalvular atrial fibrillation. J Am Heart Assoc. doi:10.1161/JAHA.116.003725
Graham DJ, Reichman ME, Wernecke M et al (2015) Cardiovascular, bleeding, and mortality risks in elderly Medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation 131:157–164
Hernandez I, Baik SH, Piñera A, Zhang Y (2015) Risk of bleeding with dabigatran in atrial fibrillation. JAMA Intern Med 175:18–24
Acknowledgements
This analysis was funded by Bristol-Myers Squibb and Pfizer. Part of this work has been presented at the European Society of Cardiology Congress 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Professor Hohnloser has served as a consultant for Bayer, BMS/Pfizer, Boehringer Ingelheim, Daiichi Sankyo, and Jansen. Professor Nabauer has received lecture fees from Bayer, BMS/Pfizer, Boehringer Ingelheim, and Daiichi Sankyo. Dr. Basic is an employee of Pfizer Deutschland GmbH. The authors have indicated that they have no other conflicts of interest regarding the content of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hohnloser, S.H., Basic, E. & Nabauer, M. Comparative risk of major bleeding with new oral anticoagulants (NOACs) and phenprocoumon in patients with atrial fibrillation: a post-marketing surveillance study. Clin Res Cardiol 106, 618–628 (2017). https://doi.org/10.1007/s00392-017-1098-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-017-1098-x