
Abstract
Heart rate influences myocardial oxygen demand, coronary blood flow, and myocardial function. Clinical and experimental studies support an association between elevated resting heart rate and a broad range of maladaptive effects on the function and structure of the cardiovascular system. Heart rate has been shown to be an important predictor of mortality in cardiovascular disorders such as coronary artery disease, myocardial infarction, and chronic heart failure. This review summarizes the specific influence of heart rate on vascular morphology and function as well as on myocardial lesions leading from early impact on vascular homeostasis to myocardial hemodynamics in chronic heart failure. Heart rate can be easily determined during physical examination of the patient and therefore allows a simple hint on prognosis and efficiency of therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Clinical background
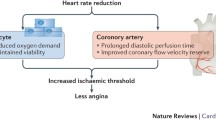
Heart rate is a major determinant of myocardial oxygen demand, coronary blood flow, and myocardial performance and affects nearly all stages of cardiovascular disease (Fig. 1) [1, 2]. In the past decades an extensive set of investigations focussed on the predictive value of heart rate for the general population and individuals affected by cardiovascular injuries [3]. The recent literature indicates that an elevated resting heart rate represents a cardiovascular risk factor, independent of currently accepted risk factors and other potentially confounding demographic and physiological characteristics [4–7]. Elevated resting heart rate has been shown to be an important predictor of mortality in cardiovascular disorders such as coronary artery disease, myocardial infarction, and chronic heart failure [8–10].

Heart rate and cardiovascular continuum
Experimental and clinical evidence suggest that sustained elevations in heart rate—irrespective of the underlying trigger—play a direct role in the pathogenesis of atherosclerosis and myocardial injuries, affect initiation and progression as well as the severity of the disease, and contribute to precipitation of vascular and myocardial events. The following sections summarize the specific role of heart rate on vascular morphology and function as well as on myocardial lesions leading from early impact on vascular homeostasis to myocardial hemodynamics in chronic heart failure. The data suggest that heart rate represents a link between cardiovascular pathology and therapy in clinical praxis.
Endothelial dysfunction
Endothelial dysfunction has been identified as an integrating pathologic phenomenon of all cardiovascular risk factors and plays a pivotal role in the development, progression, and clinical manifestations of atherosclerotic disease [11]. The clinical relevance of a connection between heart rate and endothelial integrity becomes evident in individuals with apparent microalbuminuria. Microalbuminuria defined as a urinary albumin excretion (UAE) of 30–300 mg/l delineates generalized endothelial injury and dysfunction, and several studies suggest that microalbuminuria can be accepted as an indicator of renal and cardiovascular end organ damage [12–14]. Data from the I-SEARCH study showed that heart rate is a strong predictor for the prevalence of microalbuminuria even after adjustment for hypertension and risk factors such as pre-existing cardiac disease, diabetes, age, and gender [15]. Furthermore, the positive association was aggravated in patients with a history of atrial fibrillation (AF), as prevalence of microalbuminuria at the same heart rate was about 10% higher, when compared with patients without AF [16].
A significant correlation between heart rate and endothelial function was shown in mice models of lipid-induced atherosclerosis. Oral treatment with ivabradine, a selective inhibitor of the I(f)-channel in the sinoatrial node, reduced heart rate by ~13% which was associated with improved endothelial-dependent vasorelaxation [17]. In dyslipidaemic mice, expressing the human ApoB-100, heart rate reduction with ivabradine (~17%) prevented lipid-induced endothelial dysfunction in renal and cerebral arteries [18]. One of the major underlying mechanisms that contribute to the restoration of endothelial function by heart rate reduction was shown to be a decrease of vascular oxidative stress. Consistent with these results, heart rate reduction by ivabradine improves murine penile endothelial function by reduction of oxidative stress and penile fibrosis in ApoE-knockout mice [19].
Atherosclerosis
The role of heart rate for the susceptibility of the vasculature to atherosclerosis has been extensively investigated in animal models. Early experimental studies established the causal connection between heart rate and lipid-induced atherogenesis in primates. In cynomolgus monkeys, where heart rate was reduced by ablation of the sinoatrial node, the extent of coronary and carotid atherosclerosis was reduced in comparison to sham-operated littermates [20, 21]. Kaplan et al. demonstrated that naturally occurring differences in casual heart rate in monkeys were related to coronary atherosclerosis. Monkeys with higher heart rate exhibited atherosclerotic lesions more than twice as extensive as low heart rate littermates [22]. In another study, the same authors reported a significant association between the extent of heart rate response to psychologic stress and the degree of coronary atherosclerosis in monkeys [23]. Korshunov and Berk characterized carotid outward remodeling and intima media thickening (IMT) in different inbred mouse strains. Vascular remodeling was highly dependent on genetic determinants and hemodynamic factors [24]. Among those specifically heart rate, but not systolic blood pressure, was predictive for increased IMT.
Recently the effect of heart rate reduction by ivabradine in ApoE−/− mice was characterized. Ivabradine decreased atherosclerotic plaque size in the aortic root and in the ascending aorta [17]. In another study, two different treatment regimens were applied to differentiate between preventive and curative effects of heart rate reduction on atherogenesis in ApoE−/− mice. In a preventive approach, mice were treated with ivabradine simultaneously with a high-cholesterol diet whereas in a curative intention animals were fed a high-cholesterol diet for 4 weeks to induce atherosclerosis. After 4 weeks treatment with ivabradine was started and maintained for 3 months. In both treatment regimens heart rate reduction by ivabradine significantly reduced atherosclerotic lesion formation in the aortic root [19]. Further experiments characterized potential effectors sensitive to heart rate that link impairment of endothelial homeostasis to atherosclerotic lesion formation. Selective heart rate reduction led to a reduction in monocytes chemoattractant protein-1 (MCP-1) mRNA expression. However, heart rate reduction had no effect on the aortic mRNA expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). Further research is needed to understand the molecular details of these observations.
Perski and colleagues [25, 26] studied the progression of coronary artery lesions in men who survived a myocardial infarction before the age of 45 years and underwent subsequent coronary angiographies and reported a significant correlation between heart rate and the severity and progression of coronary atherosclerosis. However, whereas animal studies provide evidence for a significant association between heart rate and an atherosclerotic phenotype, clinical data are limited.
Plaque disruption
In addition to the involvement in atherogenesis heart rate may add to the hemodynamic triggers that affect lesion texture and stability and facilitate the underlying pathology initiating coronary vascular events such as coronary plaque disruption. In a retrospective analysis, Heidland et al. investigated data from 106 patients who underwent two coronary angiographies 6 months apart. They analyzed 53 patients with initially smooth stenotic lesions who developed plaque disruption by the time of the second coronary angiogram and compared these patients with matched individuals exhibiting smooth stenoses without angiographic signs of plaque disruption. Logistic regression analysis identified positive associations between plaque disruption and a mean HR of more than 80 bpm and a negative association with the use of β-blocker [27].
The relation between atherosclerosis progression and HR can be partially explained by a dynamic coronary geometry and alternating dynamic changes imposed on the vessel during the cardiac cycle. It is recognized that in addition to established risk factors hemodynamic and wall mechanical forces are involved in the initiation and development of coronary atherosclerosis [28]. The motion of the coronary arteries during the cardiac cycle—primarily of the epicardial segments—is characterized by phasic bending of the curvatures and periodically changing torsion of the vessel directly affecting hemodynamic properties [28]. Yang et al. [29] applied computational MRI-based models to characterize mechanical stress imposed on coronary atherosclerotic plaques and identified cyclic bending as a relevant stressor in rupture-prone areas. Other types of mechanical stress affecting plaque morphology are circumferential wall stress and repetitive tensile stress which have been shown to facilitate and to trigger coronary plaque rupture [30]. Tensile stress was shown to upregulate matrix metalloproteinases—key players in extracellular matrix degradation and plaque rupture—in coronary artery lesions [31]. Lee et al. investigated mechanical properties of explanted human aortic plaques and related the stiffness of fibrous caps to histological structure and varying frequencies of radial stresses applied on the plaque. Stiffness of the plaque caps was strongly related to the frequency of applied stress and was augmented with increased frequency [32]. Consistently, all hemodynamic factors that may precipitate plaque disruption are characterized by pulsatility and frequency of mechanic stress during the cardiac cycle. However, further investigations are needed to fully understand the underlying pathology.
Coronary artery disease and myocardial infarction
The long-term prognosis of patients with stable coronary artery disease is correlated with the resting heart rate [26]. Diaz et al. [9] found in a study of 24,913 patients that the total mortality rate, the mortality rate of cardiovascular diseases as well as the rate of cardiovascular rehospitalisations increases with increasing heart rate. Patients with a resting heart rate of more than 83 bpm had an increased relative risk of 1,24 and an elevated cardiovascular mortality risk of 1,31 compared with the control group. Myocardial infarction develops when coronary plaques rupture and thrombosis occludes the vessel. Several trials demonstrated the relevance of heart rate for the prognosis of patients after myocardial infarction. According to Hjalmarson et al. [8] heart rate of patients with myocardial infarction was significantly higher than that of the control group. Furthermore, higher heart rates of these patients at hospital discharge correlate with an increase in mortality rate after 1 year (Fig. 2). Metaanalyses of the GISSI-2 and GISSI-3 trials including about 20,000 patients demonstrated that in hospital mortality rate of patients after myocardial infarction rises from 3.3 to 10.1% when patients with heart rates <60 bpm were compared with those with heart rates >100 bpm on admission [33].

Relation of between resting heart rate and survival in post-myocardial infarction patients. Redrawn from Hjalmarson et al. [8]
The GISSI-Trials showed that even patients without heart failure and an elevated heart rate had a worse long-term survival prognosis [33]. The relevance of heart rate after myocardial infarction is supported by β-blocker trials. Heart rate reduction by β-blockers is associated with a decrease in total mortality and sudden cardiac death (Fig. 3) [34–37]. In addition, heart rate-reducing verapamil-like calcium antagonists exhibited beneficial effects on prognosis of patients after myocardial infarction in the absence of heart failure [38]. In contrast, dihydropyridine-type calcium antagonists did not confer positive effects on survival which can be explained by reflex tachycardia.

Relation between reduction of resting heart rate caused by β- or calcium channel blockers and all-cause mortality in patients post myocardial infarction. Tertile subgroup analysis and meta regression of randomized clinical trials. Odds ratios represented are comparing the odds between active treatment and placebo group. Redrawn from Cucherat [34]
The first important mortality and morbidity study on pure heart rate reduction in patients with heart failure and coronary artery disease was recently published by the BEAUTIFUL (morBidity- mortality EvAlUaTion of the I(f)-inhibitor ivabradine in patients with coronary artery disease and left ventricULar dysfunction) investigators [11]. This international, multicentric trial was designed as randomized, double-blind, placebo-controlled investigation with 10917 patients enrolled who were afflicted with coronary artery disease and left ventricular dysfunction (EF <40%). The aim of the study was to investigate whether lowering heart rate with ivabradine reduces cardiovascular death and morbidity in these patients. The intervention group was treated with 5 mg b.i.d. of ivabradine with a target dose of 7.5 mg b.i.d. The patients continued therapy with ACE-I- and ARB- agents (90% of patients) as well as β-blockers (87%). The mean heart rate at entry was rather low (71.6 bpm), the mean ejection fraction of LV (32.4%) was significantly reduced. The primary endpoint comprised a composite of cardiovascular death, admission to hospital for acute myocardial infarction, admission to hospital for new onset or worsening of heart failure and revascularization. The patients were investigated by intention to treat strategy.
The results of the trial confirmed that a resting heart rate >70 bpm is associated with cardiovascular events [10]. Patients with elevated heart rates (mean 79.2 bpm) bear higher risks for coronary revascularization (+38%), admission to hospital for myocardial infarction (+46%) or heart failure (+53%), and cardiovascular death (+34%) compared with those with lower heart rates (64.1 bpm) [10]. However, treatment with ivabradine did not reduce the primary composite endpoint compared with standard medical therapy. In the subgroup of patients with baseline heart rate higher than 70 bpm (mean 79.2 bpm) ivabradine decreased the risk for fatal and non- fatal acute myocardial infarction (−36%), and the risk of coronary revascularization (−30%) compared with placebo. In contrast to these beneficial results of the subgroup analysis for the vascular endpoint, ivabradine did not improve the morbidity associated with heart failure.
Other studies demonstrated that ivabradine in combination with β-blocker treatment improved exercise tolerance, reduced angina pectoris attacks and nitro consumption in patients with stable angina compared with single β-blocker treatment [39, 40].
Heart failure
Elevated heart rate at rest is one of the key findings in acute and chronic heart failure. Heart failure has been characterized as a disorder in which the ventricles fail to provide a sufficient cardiac output to meet the requirements of the peripheral organs at normal filling pressures. Recently, it has been described that this syndrome is more heterogeneous and consists of heart failure associated with reduced ventricular ejection fraction (HFREF, systolic heart failure) as well as heart failure in combination with preserved ejection fraction (HFPEF). The hallmark of HFREF is a loss or dysfunction of myocardial muscle cells resulting in reduced systolic function. In contrast, HFPEF is often delineated by concentric left ventricular hypertrophy (wall thickening) combined with a normal or mildly abnormal cardiac ejection fraction (>50%). The hemodynamic state of the latter disease is determined by impaired diastolic relaxation and compliance sometimes together with an abnormal ventricular systolic and arterial stiffness [41]. These disorders impair ventricular filling with a concomitant increase in end-diastolic filling pressures.
In HFREF patients, large-scale randomized and controlled mortality and morbidity studies on treatment with β-blockers, ACE inhibitors (ACE-I), angiotensin receptor blockers (ARB), and aldosterone antagonists (AA) have demonstrated beneficial effects on mortality and morbidity by combination of these agents.
The significance of heart rate on mortality was retrospectively addressed in subanalyses of CIBIS II (bisoprolol; 43), MERIT-HF (metoprolol; 44), and the COMET trial (carvedilol/metoprolol; 45). These large studies enrolled together a total of almost 10,000 patients with advanced systolic heart failure (NYHA class II–IV). The general trend of these three trials clearly demonstrates that high heart rate at rest contributes to poor survival. These experiences are in line with previous studies of β-blockers and other drugs approved in heart failure therapy, demonstrating greater benefits with higher baseline heart rates (>80 bpm) as well as markedly reduced heart rates (>10 bpm) after drug treatment [42]. However, it remains currently unknown whether or to what extent the benefit from β-blockers in patients with heart failure is due to heart rate reduction per se or other beneficial effects derived from interrupting maladaptive β-signaling pathways (apoptosis, fetal gene expression, Ca2+ handling, etc.). A recent metaanalysis aimed at determining whether the survival benefits of β-blockade in heart failure is associated to the magnitude of heart rate reduction or β-blocker dose showed a close relation of outcome with the former, but not the latter [43]. As β-blockers considerably reduce heart rate (negative chronotropy) they are most appropriate to be compared with ivabradine [44, 45].
If ivabradine should decrease mortality and morbidity in heart failure, baseline rate (>80 bpm) and the extent of heart rate reduction (−10 to 20 bpm) should probably be in the same order of magnitude as in the large β-blocker trials [46–48]. The “Systolic heart failure treatment with the I(f)-inhibitor ivabradine Trial” (SHIFT) fulfilled these criteria. The aim of the “SHIFT” was to evaluate the effect of pure heart rate reduction by ivabradine in addition to guideline-based treatment on cardiovascular outcomes, symptoms and quality of life in patients with systolic heart failure [49]. This international, multicentric trial was designed as randomized, double-blind, placebo-controlled investigation with 6,505 patients enrolled who were afflicted with left ventricular dysfunction (EF ≤ 35%), sinus rhythm and symptomatic heart failure of more than 4 weeks duration (mainly NYHA Stadium II-III). The intervention group was treated with an average dose of 6.4 mg b.i.d. of ivabradine on top of standard therapy with ACE-I and ARB agents (about 92% of patients) as well as β-blockers (90%). Similar to the β-blocker trials, treatment with ivabradine was associated with an average reduction of heart rate of 15 bpm from a baseline value of 80 bpm, which was mainly maintained throughout the course of the trial. Heart rate reduction by ivabradine reduced the risk for the primary endpoint of cardiovascular death or hospital admission for worsening of heart failure in the Shift study population by 18%. Furthermore, patients with heart rates higher than the mean value (80 bpm) had an increased risk for adverse effects and received greater benefit from ivabradine treatment than those patients with heart rates lower than the median. A further analysis of the SHIFT data demonstrated, that in patients with systolic heart failure, a continuous relationship between baseline heart rate and adverse outcomes existed. The risk was modified and significantly decreased by heart rate reduction via ivabradine [50]. Therefore, SHIFT demonstrated for the first time beneficial effects of heart rate reduction alone in patients with systolic heart failure.
The relatively low baseline heart rate (mean 71.6 bpm) and the small reduction in heart rate (mean 6 bpm) in the BEAUTIFUL study by ivabradine may be an explanation why ivabradine in this study failed to demonstrate improvement in heart failure-related outcomes in patients with stable angina and left ventricular dysfunction [11]. These results may indicate that baseline heart rate possibly due to preexisting β-blockade was too low that I(f)-channel inhibition was unable to gain further therapeutics impact in these patients. Another explanation may be that the background cardiac conditions differed in both study populations.
In heart failure survival of HFREF patients unequivocally improved with the currently recommended therapy, whereas survival rates of HFPEF patients have remained rather unchanged during the past two decades [51]. Current therapy is only symptomatic. Apart from prescribing diuretics for HFPEF patients with pulmonary congestion and edema, therapeutic goals of these disease entities are treatment of hypertension and myocardial ischemia, maintenance of sinus rhythm, optimal treatment of diabetes mellitus, and heart rate control. Beside of lowering afterload, heart rate reduction prolongs diastole and LV filling time providing additional time for diastolic relaxation due to improved Ca-ion reuptake into the sarcoplasmic reticulum. Consequently, lower heart rate reduces pulmonary pressure and extends diastolic coronary perfusion time, thereby preventing ischemia-induced diastolic dysfunction [52].
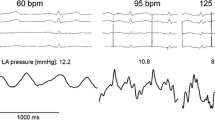
A special case of unloading of the left ventricle is elegantly demonstrated in a patient with HFPEF by heart rate reduction (Fig. 4). In the upper panel left ventricular and pulmonary artery pressure curves at high heart rate (119 bpm) are depicted. After an injection of 5 mg metoprolol the heart rate decreases (96 bpm) at a nearly constant systemic pressure (aorta) while the pressure of pulmonary artery considerably declines (lower panel). These findings demonstrate that improvement of LV diastolic filling of this HFPEF patient is associated with a decrease in pulmonary pressure. It would therefore be of interest to test in a clinical study whether heart rate reduction by ivabradine would be equal or even superior to β-blockers in this special hemodynamic setting. At present, no data with specific heart rate reducing agents are available in HFNEF patients.

Hemodynamic effects induced by lowering heart rate in a patient with HFPEF a LV pressure (red pressure curve) and pulmonary artery pressure (white pressure curve) at high heart rate (120 bpm). b After intravenous administration of 5 mg metoprolol heart rate is reduced (96 bpm). As a result the mean pressure of the mean pulmonary artery considerably declined (from 46 to 27 mmHg) while systemic pressure (aorta) remained virtually unchanged
Individual experiences with ivabradine medication can be drawn from case reports dealing with special hemodynamic alterations in cardiovascular disease, in particular in heart failure [53]. A HFREF-patient with acute left ventricular decompensation on adequate heart failure medication developed sinus tachycardia (120 bpm) with dyspnoe under dobutamine infusion in the cardiac care unit. Consequently, ivabradine was carefully titrated orally up to a dose of 15 mg/day to reduce heart rate under ongoing dobutamine therapy. Within in 5 days heart rate decreased (−34%) with a striking increase in stroke volume (+40%) accompanied by a simultaneously decrease in systemic and pulmonary vascular resistance (Fig. 5). After ivabradine withdrawal hemodynamic values worsened again, but re-administration of ivabradine led to weaning from dobutamine therapy in the next 3 days (Fig. 5) and the patient recovered. This report and previous studies may be helpful to generate hypotheses for future studies on the hemodynamic effects of ivabradine in acute hemodynamic deterioration [54].

Hemodynamic improvement on ivabradine medication (up to 15 mg in 5 days) during acute left ventricular decompensation in advanced heart failure. Ivabradine was titrated, withdrawn (0 mg/day), and re-administered in order to wean the patient from dobutamine infusion (9μg/kg/min). a Heart rate (HR, bpm), b stroke volume (SV, ml), c systemic vascular resistance (SVR, dyn s/cm5), d pulmonary vascular resistance (PVR, dyn s/cm5). Modified from Link et al. [53]
Several questions regarding heart failure therapy remain currently unresolved. It is important to learn to what extent resting heart rate should be reduced to gain a benefit for patients with heart failure. It is interesting to elucidate whether heart rate reduction alone or together with its specific mode of action (β-blockade, I(f)-channel inhibition, exercise training, etc.) may lead to improved outcome. It is of practical importance to realize that despite the high percentage β-blocker treatment in patients with heart failure therapy, only about one-third of the treated patients are on prescribed β-blocker dose and therefore on proper resting heart rate [55].
Conclusions
Clinical and experimental studies support an association between elevated resting heart rate and a broad range of maladaptive vascular effects. Increased heart rate impairs endothelial function in animal models and may contribute to reduced vascular function. Heart rate reduction inhibits formation of atherosclerotic plaque in animal models of lipid-induced atherosclerosis and may prevent or retard the final development in chronic heart failure. Whereas experimental data provide considerable evidence of the pathophysiological concept, transition from experimental results to clinical evidence has to be further established. It needs to be clarified whether a selective pharmacological heart rate reduction per se might be beneficial for prevention of cardiovascular disease. New clinical studies are designed and focussed on this specific topic.
References
Heusch G (2008) Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol 153(8):1589–1601
Reil JC, Böhm M (2007) The role of heart rate in the development of cardiovascular disease. Clin Res Cardiol 96(9):585–592
Levy RL, White PD, Stroud WD (1945) Transient tachycardia: prognostic significance alone and in association with transient hypertension. JAMA 129:585–588
Kannel WB, Kannel C, Paffenbarger RS Jr, Cupples LA (1987) Heart rate and cardiovascular mortality: the Framingham study. Am Heart J 113(6):1489–1494
Fox K, Borer JS, Camm AJ, Danchin N, Ferrari R, Lopez Sendon JL, Steg PG, Tardif JC, Tavazzi L, Tendera M (2007) Resting heart rate in cardiovascular disease. J Am Coll Cardiol 50(9):823–830
Graham I, Atar D, Borch-Johnsen K, Boysen G, Burell G, Cifkova R, Dallongeville J, De Backer G, Ebrahim S, Gjelsvik B, Herrmann-Lingen C, Hoes A, Humphries S, Knapton M, Perk J, Priori SG, Pyorala K, Reiner Z, Ruilope L, Sans-Menendez S, Scholte op Reimer W, Weissberg P, Wood D, Yarnell J, Zamorano JL, Walma E, Fitzgerald T, Cooney MT, Dudina A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Funck-Brentano C, Filippatos G, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Altiner A, Bonora E, Durrington PN, Fagard R, Giampaoli S, Hemingway H, Hakansson J, Kjeldsen SE, Larsen ML, Mancia G, Manolis AJ, Orth-Gomer K, Pedersen T, Rayner M, Ryden L, Sammut M, Schneiderman N, Stalenhoef AF, Tokgozoglu L, Wiklund O, Zampelas A (2007) European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 28(19):2375–2414
Palatini P (2009) Elevated heart rate: a “new” cardiovascular risk factor? Prog Cardiovasc Dis 52(1):1–5
Hjalmarson A, Gilpin EA, Kjekshus J, Schieman G, Nicod P, Henning H, Ross J Jr (1990) Influence of heart rate on mortality after acute myocardial infarction. Am J Cardiol 65(9):547–553
Diaz A, Bourassa MG, Guertin MC, Tardif JC (2005) Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J 26(10):967–974
Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R (2008) Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet 372(9641):817–821
Fox K, Ford I, Steg PG, Tendera M, Ferrari R (2008) Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind placebo-controlled trial. Lancet 372(9641):807–816
Pedrinelli R, Giampietro O, Carmassi F, Melillo E, Dell’Omo G, Catapano G, Matteucci E, Talarico L, Morale M, De Negri F et al (1994) Microalbuminuria and endothelial dysfunction in essential hypertension. Lancet 344(8914):14–18
Stehouwer CD, Henry RM, Dekker JM, Nijpels G, Heine RJ, Bouter LM (2004) Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction—the Hoorn Study. Kidney Int Suppl (92):S42–S44
Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A (1989) Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 32(4):219–226
Böhm M, Reil JC, Danchin N, Thoenes M, Bramlage P, Volpe M (2008) Association of heart rate with microalbuminuria in cardiovascular risk patients: data from I-SEARCH. J Hypertens 26(1):18–25
Böhm M, Thoenes M, Neuberger HR, Graber S, Reil JC, Bramlage P, Volpe M (2009) Atrial fibrillation and heart rate independently correlate to microalbuminuria in hypertensive patients. Eur Heart J 30(11):1364–1371
Custodis F, Baumhakel M, Schlimmer N, List F, Gensch C, Böhm M, Laufs U (2008) Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation 117(18):2377–2387
Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC (2008) Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol 154(4):749–757
Baumhäkel M, Custodis F, Schlimmer N, Laufs U, Böhm M (2010) Heart rate reduktion with ivabradine improves erectile dysfunction in parallel to decrease in atherosclerotic plaque load in ApoE-knockout mice. Atherosclerosis (Mar 9 Epub ahead of print)
Beere PA, Glagov S, Zarins CK (1984) Retarding effect of lowered heart rate on coronary atherosclerosis. Science 226(4671):180–182
Beere PA, Glagov S, Zarins CK (1992) Experimental atherosclerosis at the carotid bifurcation of the cynomolgus monkey localization, compensatory enlargement, and the sparing effect of lowered heart rate. Arterioscler Thromb 12(11):1245–1253
Kaplan JR, Manuck SB, Clarkson TB (1987) The influence of heart rate on coronary artery atherosclerosis. J Cardiovasc Pharmacol 10(Suppl 2):S100–S102 discussion S103
Manuck SB, Adams MR, McCaffery JM, Kaplan JR (1997) Behaviorally elicited heart rate reactivity and atherosclerosis in ovariectomized cynomolgus monkeys (Macaca fascicularis). Arterioscler Thromb Vasc Biol 17(9):1774–1779
Korshunov VA, Berk BC (2004) Strain-dependent vascular remodeling: the “Glagov phenomenon” is genetically determined. Circulation 110(2):220–226
Perski A, Hamsten A, Lindvall K, Theorell T (1988) Heart rate correlates with severity of coronary atherosclerosis in young postinfarction patients. Am Heart J 116(5 Pt 1):1369–1373
Perski A, Olsson G, Landou C, de Faire U, Theorell T, Hamsten A (1992) Minimum heart rate and coronary atherosclerosis: independent relations to global severity and rate of progression of angiographic lesions in men with myocardial infarction at a young age. Am Heart J 123(3):609–616
Heidland UE, Strauer BE (2001) Left ventricular muscle mass and elevated heart rate are associated with coronary plaque disruption. Circulation 104(13):1477–1482
Zhu H, Friedman MH (2003) Relationship between the dynamic geometry and wall thickness of a human coronary artery. Arterioscler Thromb Vasc Biol 23(12):2260–2265
Yang C, Tang D, Kobayashi S, Zheng J, Woodard PK, Teng Z, Bach R, Ku DN (2008) Cyclic bending contributes to high stress in a human coronary atherosclerotic plaque and rupture risk: in vitro experimental modeling and ex vivo MRI-based computational modeling approach. Mol Cell Biomech 5(4):259–274
Katritsis DG, Pantos J, Efstathopoulos E (2007) Hemodynamic factors and atheromatic plaque rupture in the coronary arteries: from vulnerable plaque to vulnerable coronary segment. Coron Artery Dis 18(3):229–237
Lee RT, Schoen FJ, Loree HM, Lark MW, Libby P (1996) Circumferential stress and matrix metalloproteinase 1 in human coronary atherosclerosis. Implications for plaque rupture. Arterioscler Thromb Vasc Biol 16(8):1070–1073
Lee RT, Grodzinsky AJ, Frank EH, Kamm RD, Schoen FJ (1991) Structure-dependent dynamic mechanical behavior of fibrous caps from human atherosclerotic plaques. Circulation 83(5):1764–1770
Zuanetti G, Hernándes-Bernal F, Rossi A, Comerio G, Paolucci G, Maggioni AP (1999) Relevance of heart rate as a prognostic factor in myocardial infarction: the GISSI experience. Eur Heart J 1(suppl H):H52–H57
Cucherat M (2007) Quantitative relationship between resting heart rate reduction and magnitude of clinical benefits in post-myocardial infarction: a meta-regression of randomized clinicat trials. Eur Heart J 28:3012–3019
Kendall MJ, Lynch KP, Hjalmarson A, Kjekshus J (1995) Beta-blockers and sudden cardiac death. Ann Intern Med 123(5):358–367
Hjalmarson A (1997) Effects of beta blockade on sudden cardiac death during acute myocardial infarction and the postinfarction period. Am J Cardiol 80(9B):35J–39J
Beta-blocker Heart Attack Trial Research Group (1982) A randomized trial of propranolol in patients with acute myocardial infarction mortality results. JAMA 247(12):1707–1714
Danish Study Group on Verapamil in myocardial infarction (1990) Effect of verapamil on mortality and major events after acute myocardial infarction (the Danish Verapamil Infarction Trial II (DAVIT II). Am J Cardiol 66:770–785
Tardif JC, Ponikowski P, Kahan T (2009) for the ASSOCIATE study investigators. Efficacy of the I(f) current inhibitor ivabradine in patients with chronic stable angina receiving beta blocker therapy: a 4-month, randomized placebo-controlled trial. Eur Heart J 30:540–548
Koester R, Kaehler J, Ebelt H, Soeffker G, Werdan K, Meinertz T. (2010) Ivabradine in combination with beta-blocker therapy for the treatment of stable angina pectoris in every day clinical practice. Clin Res Cardiol (Epub ahead of print)
Kawaguchi M, Hay I, Fetics B, Kass DA (2003) Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation 107:714–720
Kjekshus J, Gullestad L (1999) Heart rate as therapeutic target in heart failure. Eur Heart J 1(Suppl. H):H64–H69
McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW (2009) Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med 150:784–794
Reil J-C, Reil G-H, Böhm M (2009) Heart rate reduction by I(f)-channel inhibition and its potential role in heart failure with reduced and preserved ejection fraction. Trends Cardiovasc Med 19:152–157
DiFrancesco D, Camm AJ (2004) Heart rate lowering by specific and selective I(f) current inhibition with ivabradine. A new therapeutic perspective in cardiovascular disease. Drugs 64:1757–1765
Lechat P, Hulot JS, Escolano S et al (2001) Heart rate and cardiac rhythm relationships with bisoprolol benefit in chronic heart failure in CIBIS II Trail. Circulation 103:1428–1433
Gullestad L, Wikstrand J, Deedwania P (2005) What resting heart rate should one aim for when treating patients with heart failure with a Beta-Blocker? J Am Coll Cardiol 45:252–259
Metra M, Torp-Pedersen C, Swedberg K et al (2005) Influence of heart rate, blood pressure, and beta-blocker dose on outcome and the difference in outcome between carvedilol and metoprolol tartrate in patients with chronic heart failure: results from the COMET trial. Eur Heart J 26:2259–2268
Swedberg K, Komajda M, Böhm M, Borer JS et al (2010) Ivabradine and outcomes in chronic heart failure (SHIFT): a randomized placebo-controlled study. Lancet [Epub ahead of print]
Böhm M, Swedberg K, Komajda M, Borer JS et al (2010) Heart rate as a risk factor in chronic heart failur (SHIFT): the association between heart rate and outcomes in a randomized placebo-controlled trial. Lancet [Epub ahead of print]
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355:251–259
Kindermann M, Reil J-C, Pieske B, van Veldhusen DJ, Böhm M (2008) Heart failure with normal left ventricular ejection fraction (HFNEF). What is evidence? Trends Cardiocasc Med 18:280–292
Link A, Reil JC, Selejan S, Böhm M (2009) Effect of ivabradine in dobutamine induced sinus tachycardia in a case of acute heart failure. Clin Res Cardiol 98:513–551
De Ferrari GM, Mazzuero A, Agnesina L, Bertoletti A et al (2008) Favourable effects of heart rate reduction with intravenous administration of ivabradine in patients with advanced heart failure. Eur J Heart Fail 10:550–555
Deedwania P, Carbajal E, Dietz R, Mukherjee R et al (2006) Heart rate is powerful predictor in mortality in post-AMI patients with heart failure: results from the EPHESUS trial. Eur Heart J 27:590
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reil, JC., Custodis, F., Swedberg, K. et al. Heart rate reduction in cardiovascular disease and therapy. Clin Res Cardiol 100, 11–19 (2011). https://doi.org/10.1007/s00392-010-0207-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-010-0207-x