
Abstract
Background and aims
The adenomatous polyposis coli (APC) protein plays a crucial role in the regulation of β-catenin, which is linked to the cell adhesion molecule E-cadherin. Furthermore, β-catenin and cyclooxygenase-2 (COX-2) are both involved in the activation of nuclear transcription factors inducing cell proliferation. Germline mutations in the APC gene are the cause of familial adenomatous polyposis (FAP). To characterise the expression pattern of these proteins in FAP in comparison with sporadic adenomas, we studied 18 FAP-associated adenomas, 16 sporadic adenomas and seven normal colonic controls.
Methods
E-cadherin, β-catenin, COX-2 expression and the proliferative index (Ki67) were assessed by immunohistochemistry (index of expressing cells / total number of cells) in adenomatous mucosa, adjacent non-neoplastic tissue and normal colonic controls.
Results
E-cadherin expression was significantly and homogeneously reduced in FAP adenomas (24%; 95%CI 16–32; sporadic adenomas 61%; 38–84; normal controls 98%; 96–100). Membraneous β-catenin expression was significantly reduced in both FAP (30%; 11–49) and sporadic (42%; 19–65) adenomas (normal controls 96%; 88–104), whereas marked nuclear staining occurred in sporadic, but not in FAP adenomas. Stromal COX-2 expression and the proliferative index were increased only in sporadic adenomas (sporadic adenomas: COX-2 12%; 7–17, Ki67 24%; 15–33, FAP adenomas: COX-2 8%; 5–11, Ki67 5%; 2–9, normal controls: COX-2 4%; 2–7, Ki67 6%; 1–11).
Conclusion
Proteins involved in cell adhesion and cell proliferation, especially E-cadherin, are expressed differently in FAP and sporadic adenoma, pointing to possible differences in the molecular pathways to adenoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Familial adenomatous polyposis (FAP) is caused by germline mutations in the adenomatous polyposis coli (APC) gene, which leads to the development of hundreds of colorectal adenomas. The crucial role of the APC gene for the regulation of cellular proliferation and differentiation was evaluated by Kinzler and Vogelstein in their genetic model of colorectal carcinogenesis [1]. APC interacts with β-catenin. This protein plays a pivotal role in cell proliferation and cell adhesion pathways. On the one hand, it is linked to E-cadherin, the key protein for cell-cell-adhesion in adherens junctions. E-cadherin is anchored to the cytoskeleton: the cytoplasmic tail of E-cadherin binds to β-catenin, which binds to α-catenin, which—in turn—interacts with actin filaments [2] and β-catenin. The interruption of the E-cadherin mediated cell adhesion is associated with invasiveness of tumour cells [3]. On the other hand, β-catenin is a key regulator of cellular proliferation. The level of free cytosolic β-catenin is regulated under the influence of the Wnt proteins by a protein degradation complex containing APC. Unbound cytosolic β-catenin can translocate to the nucleus and bind to proteins of the lymphocyte enhancer factor (LEF) / T cell factor (TCF) protein family [4]. Their target genes, e.g. cyclin D1 and the peroxisome proliferator-activated receptor δ(PPARδ) gene [5, 6, 7], are involved in cell cycle control. In colorectal tumours this pathway is activated by two alternative mechanisms: either the interaction between APC and β-catenin is compromised due to a mutation of the APC gene or β-catenin expression is increased due to a mutation in the β-catenin gene (CTNNB1) itself [8].
Recently, alternative pathways to colorectal neoplasia have been described: in contrast to classical sporadic adenomas, alterations of APC do not appear to be an early event in the development of so called serrated adenomas. Instead, resistance to apoptosis and a defective DNA repair are the postulated hallmarks of this pathway (reviewed in [9]).
Weak or absent expression of E-cadherin, together with nuclear β-catenin expression and increased COX-2 expression, has repeatedly been noted in sporadic colorectal carcinoma as well as in other gastrointestinal neoplasms [10]. In order to further elucidate the role of these proteins in familial as well as sporadic adenoma formation, we studied the expression of E-cadherin, β-catenin, COX-2 and the proliferation marker Ki67 in biopsies taken from adenomatous and non-adenomatous mucosa of patients with FAP and sporadic adenoma in comparison with normal controls.
Methods
Patients
Eighteen adenomas taken from 18 patients with FAP during colonic surgery or colonoscopy were included in this study (mean age at diagnosis due to symptoms was 30.7 years, range 16–44 years). The diagnosis of FAP was based on the presence of more than 100 colonic adenomatous polyps (n=8), less than 100 adenomatous polyps together with an identified germline mutation in the APC gene (n=8), or colonic adenomatous polyps in combination with a positive familial history (n=2). Overall, germline mutations were known in 13/18 (72%) patients (Table 1). Sixteen adenomas from patients without FAP (single adenoma in elderly patients, mean age 62.1 years, range 52–84 years) and biopsies from seven patients without colorectal neoplasia (mean age 51.1 years, range 22–92 years) served as controls. The clinical data of the adenomas studied are summarised in Table 2. The study was approved by the Bonn University ethics committee in accordance with the Helsinki declaration of 1975, revised 1983. Each patient had given informed consent.
Genetic studies
Genomic DNA was isolated from peripheral blood lymphocytes using standard salting out techniques [11]. Screening for germline mutations was performed by nonradioactive single-strand conformation analysis (SSCP) and heteroduplex analysis with genomic DNA as described previously [12]. Alternatively, total RNA was extracted from EDTA blood samples with TRIZOL (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s protocol, and a protein truncation test (PTT) was performed using previously described primers [13]. Sequencing of aberrant PCR fragments was done with Sequenase version 2.0 (Amersham Biosciences, Freiburg, Germany) in the presence of 35S-dATP, or by semiautomatic cycle sequencing with BigDye chain terminators on an ABI 377 sequencer (Perkin Elmer, Weiterstadt, Germany).
Immunohistochemistry
Paraffin-embedded tissue was cut into 6-µm sections. Slides for E-cadherin, β-catenin and Ki67 staining were processed in citrate buffer (pH 6.0) in a microwave oven for 30 min to enhance antigen retrieval. Immunohistochemical staining was carried out with the avidin-biotin-peroxidase-complex method using 3-amino-9-ethyl-carbazole as substrate [14]. The following murine monoclonal antibodies were applied as primary antibodies: E-cadherin (clone 36, cat. no. C20820, Transduction Laboratories, Lexington, USA) diluted 1:40, β-catenin (clone 14, cat. no. C19220, Transduction Laboratories) diluted 1:160, Ki67 (clone Ki-S5, cat. no. 1742345, Roche Diagnostics, Mannheim, Germany) diluted 1:80 and COX-2 (cat. no. C22420, Transduction Laboratories) diluted 1:160.
For semiquantitative evaluation of protein expression, we identified the region of the slide with the predominant expression pattern and chose three crypts. Whenever possible, only orthogonally cut crypts were considered to ensure that only complete crypts were counted and no bias towards superficial or basal crypt cells occurred. The cells expressing the antigen in a typical fashion (E-cadherin/β-catenin: membraneous staining, COX-2: cytoplasmic staining, Ki67: nuclear staining) were counted under a microscope. The ratio of antigen-positive cells to the total number of counted cells was determined. To quantify COX-2 expression in stroma cells, three stroma areas, each limited by two crypts, were evaluated. The stained cells were counted in a high power field at a 400× magnification. Thereafter, the ratio of stained cells to the total number of counted cells was calculated. The evaluation was done by two independent observers (MJ, FG), who were not informed of the group allocation of the specimens. In the rare case of differences between the results of the two observers, the specimens were reevaluated by the two observers together and consensus was obtained.
Statistical analysis
Expression ratios in FAP-associated adenoma, sporadic adenoma and normal controls were compared by the Mann-Whitney U test for unpaired samples. All calculations were done with the SPSS software package (SPSS, Chicago, USA). Results are given as means and 95% confidence intervals. A p value <0.05 was considered to indicate statistical significance.
Results
When conventional criteria for the classification of adenoma were applied to the adenomas of our two groups of patients, the groups differed only in the size of the adenomas (Table 2). In normal mucosa, membraneous expression of E-cadherin and β-catenin was found in almost every colonic epithelial cell (Fig.1A, B). COX-2 expression was absent in epithelial cells but occasionally noted in stromal cells beneath the epithelial layer (Fig.1C). The percentage of proliferating cells as assessed by Ki67 expression was rather low (6%, Fig.1D). Table 3 summarises the semiquantitative data of all studied proteins.

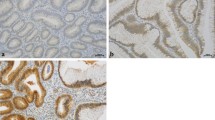
Protein expression in mucosa of normal controls. A Membraneous E-cadherin expression in all epithelial cells. B Membraneous β-catenin expression in all crypt cells. C Scarce COX-2 expression in stromal cells. Detail of the region marked in the overview by a black box. Cytoplasmic expression of COX-2 protein (small arrows). D Seldom Ki67 expression in normal mucosa. The detail is taken from the region which is marked in the overview by a black box. Nuclear staining of Ki67 (small arrow) (magnification: Overview 200×, detail: 400×)
In FAP patients, a marked reduction of E-cadherin expression was noted in the epithelial cells of adenomatous mucosa (Fig. 2). As can be seen in the semiquantitative evaluation (Table 3), E-cadherin expression in sporadic adenoma was reduced to a lesser but still significant extent. The normal mucosa adjacent to FAP-associated adenomas showed a moderate reduction in the number of cells positive for E-cadherin. This lower E-cadherin expression was comparable to the reduction found in sporadic adenoma. The numbers of E-cadherin positive cells were lower in both groups of patients with adenoma than in normal controls.

E-cadherin expression in mucosa of an FAP patient: normal membraneous expression in non-neoplastic regions, lost expression in adenomatous glands. This fact is presented in detail (normal mucosa: large arrow, adenomatous mucosa: small arrow). The region of the detail is marked by a black box (magnification: overview 40×, insert 200×)
In adenomatous mucosa of patients with sporadic and FAP-associated adenoma, membraneous β-catenin expression was reduced to a variable extent (Fig. 3A). Instead, cytoplasmic and nuclear β-catenin immunoreactivity was detected (Fig. 3B, C, Table 4).

β-catenin expression. A Reduction of membraneous expression (insert, arrow a) and appearance of cytoplasmic (insert, arrow b) and nuclear (insert, arrow c) β-catenin expression in a sporadic adenoma (magnification: overview 40×, insert 400×). B Widespread nuclear expression in a sporadic adenoma (magnification 200×). C Marked cytoplasmic expression (detail: arrow) in an adenoma of an FAP patient (magnification: overview 40×, insert 400×)
Epithelial cell proliferation and stromal COX-2 expression in adenomatous mucosa of FAP patients did not reveal significant differences compared with normal controls. In contrast, both were markedly increased in sporadic adenoma (Figs. 4A and 5). It is noteworthy, however, that in adenomatous mucosa of both sporadic and FAP-associated adenoma, COX-2 expressing cells could be detected not only in stromal cells but also in epithelial cells (Fig. 4B).

COX-2 expression. A Increased stromal COX-2 expression in a sporadic adenoma. Note the subepithelial localisation of the protein in the adenoma (arrows). Detail: documentation of the cytoplasmic expression in the region marked in the overview by a black box (magnification: overview 40×, insert 400×). B Epithelial COX-2 expression in an FAP-associated adenoma. The box marked crypt is presented in detail to demonstrate cytoplasmic expression (magnification: overview 40×, insert 400×)

Ki67 expression. A Widespread nuclear expression in crypts of a sporadic adenoma representing the increased proliferative index (magnification: 200×). B Seldom Ki67 expression in an FAP-associated adenoma. Detail: nuclear localisation of the protein (arrows) (magnification: overview 100×, insert 400×)
Discussion
The key result of this study is the highly significant and homogeneous reduction of E-cadherin expression in FAP-associated adenomas.
While the considerable heterogeneity with respect to E-cadherin expression in sporadic adenomas is compatible with previous findings [15, 16], the uniform reduction of E-cadherin expression in FAP-associated adenoma was unexpected. Reduction of E-cadherin expression is usually considered a late event in the process of gastrointestinal carcinogenesis. In sporadic colorectal carcinoma and cholangiocellular carcinoma, for example, the proportion of tumours showing reduced E-cadherin expression was higher in less-differentiated carcinomas than in well-differentiated tumours [10, 17]. Due to this fact, one might suggest that the differences between the adenoma groups could be due to different sizes. The 16 sporadic adenomas were almost twice as large as the FAP-associated adenomas. However, the difference in E-cadherin expression cannot be explained by different adenoma size since large adenomas should represent a more advanced stage of dedifferentiation [18] with less E-cadherin expression. The opposite was observed in our study.
In contrast to our results, previous studies showed considerable abnormal β-catenin expression [19] and abnormal β-catenin expression in concert with preserved membraneous E-cadherin expression [20] in FAP-associated adenomas. In both studies, only clinically characterised patients with a classical FAP phenotype were included. In our study, in most of the patients a APC germline mutation was identified, and patients with an atypical phenotype were also included. El-Bahrawy et al. reported a general overexpression of E-cadherin and β-catenin as a result of abnormal protein expression in addition to a preserved membraneous protein localisation. This unusual expression pattern has not been observed before. In addition, these data have to be questioned since an increased cellular pool of unbound β-catenin is thought to induce proliferation but there is evidence that FAP adenomas are more due to decreased apoptosis than to increased proliferation (see below). Unfortunately, El-Bahrawy’s group did not provide data on cell proliferation. Cell culture data on the interaction between E-cadherin and β-catenin indicate that abnormal β-catenin localisation in the cytoplasm disturbs E-cadherin function and may also shift the expression pattern of E-cadherin from membraneous to cytoplasmic [21]. But the reduction of E-cadherin in our FAP group can not be explained by this mechanism, because we did not observe significant cytoplasmic β-catenin and E-cadherin expression in FAP adenomas. We do not believe that the differences between our study and others’ work concerning β-catenin expression is due to methodological issues since our data on β-catenin expression in sporadic adenoma were in line with previous studies [22]. Mutations in the APC gene can lead to accumulation of β-catenin in the cytosol and translocation of β-catenin to the nucleus [23, 24]. In agreement with this concept we found nuclear expression of β-catenin in 50% of the sporadic adenomas.
The observed proliferative activity in our samples is well compatible with our β-catenin data. FAP adenomas showed proliferation rates equal to normal controls in line with the rare observation of nuclear β-catenin expression in FAP adenomas. Normal epithelial proliferation rates were also reported in the MIN mouse, a murine model for FAP. On the other hand, apoptosis was decreased in this animal model [25] and in human tissue [26]. Thus, the initial adenoma formation in FAP may be the result of decreased apoptosis rather than increased proliferation.
The more heterogeneous alteration of the expression of E-cadherin and β-catenin in sporadic adenoma may reflect the greater genetic heterogeneity of sporadic adenoma. In particular, sporadic adenoma may have additional lesions in the Wnt-pathway or may have mutations in the β-catenin (CNTTB) gene itself, postulated by cell culture data [8, 27].
COX-2 overexpression was demonstrated in patients with sporadic colorectal carcinoma and adenoma [28, 29]. Indeed, we found an increased COX-2 expression in sporadic adenoma. However, COX-2 expression was predominantly found in stroma cells directly beneath the epithelial layer rather than in epithelial cells. This expression pattern suggests a paracrine mechanism for the released COX-2 products such as prostaglandins, as already proposed by previous authors [28]. In contrast, COX-2 expression in colorectal carcinoma was observed primarily in epithelial cells. In a subset of our sporadic and FAP related adenomas, we also observed epithelial COX-2 expression.
Despite the small study population—FAP is a rare disease—we found highly significant differences between FAP and non-FAP adenomas, especially with respect to the expression of E-cadherin. The mechanism of this differences remains to be elucidated. Our results may be an additional indicator that FAP is not simply an inherited version of colorectal neoplasia but a distinct neoplasia.
References
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87:159–170
Knudsen KA, Soler AP, Johnson KR, Wheelock MJ (1995) Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol 130:67–77
Behrens J, Mareel MM, Van Roy FM, Birchmeier W (1989) Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol 108:2435–2447
Bienz M, Clevers H (2000) Linking colorectal cancer to Wnt signaling. Cell 103:311–320
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 96:5522–5527
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398:422–426
He TC, Chan TA, Vogelstein B, Kinzler KW (1999) PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 99:335–345
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275:1787–1790
Jass JR, Whitehall VLJ, Young J, Leggett BA (2002) Emerging concepts in colorectal neoplasia. Gastroenterology 123:862–876
Van Aken J, Cuvelier CA, De Wever N, Roels J, Gao Y, Mareel MM (1993) Immunohistochemical analysis of E-cadherin expression in human colorectal tumours. Pathol Res Pract 189:975–978
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Mandl M, Paffenholz R, Friedl W, Caspari R, Sengteller M, Propping P (1994) Frequency of common and novel inactivating APC mutations in 202 families with familial adenomatous polyposis. Hum Mol Genet 3:181–184
Bala S, Kraus C, Wijnen J, Meera Khan P, Ballhausen WG (1996) Multiple products in the protein truncation test due to alternative splicing in the adenomatous polyposis coli (APC) gene. Hum Genet 98:528–533
Hsu SM, Raine L, Fanger H (1981) Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 29:577–580
Gagliardi G, Kandemir O, Liu D, Guida M, Benvestito S, Ruers TG, et al (1995) Changes in E-cadherin immunoreactivity in the adenoma-carcinoma sequence of the large bowel. Virchows Arch 426:149–154
Valizadeh A, Karayiannakis AJ, el-Hariry I, Kmiot W, Pignatelli M (1997) Expression of E-cadherin-associated molecules (alpha-, beta-, and gamma-catenins and p120) in colorectal polyps. Am J Pathol 150:1977–1984
Ashida K, Terada T, Kitamura Y, Kaibara N (1998) Expression of E-cadherin, alpha-catenin, beta-catenin, and CD44 (standard and variant isoforms) in human cholangiocarcinoma: an immunohistochemical study. Hepatology 27:974–982
Jorgensen OD, Kronborg O, Fenger C (1993) The Funen Adenoma Follow-Up Study. Characteristics of patients and initial adenomas in relation to severe dysplasia. Scand J Gastroenterol 28:239–243
Inomata M, Ochiai A, Akimoto S, Kitano S, Hirohashi S (1996) Alteration of beta-catenin expression in colonic epithelial cells of familial adenomatous polyposis patients. Cancer Res 56:2213–2217
El-Bahrawy MA, Talbot IC, Poulsom R, Jeffery R, Alison MR (2002) The expression of E-cadherin and catenins in colorectal tumours from familial adenomatous polyposis patients. J Pathol 198:69–76
Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, et al (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120:757–766
Kobayashi M, Honma T, Matsuda Y, Suzuki Y, Narisawa R, Ajioka Y, et al (2000) Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer 82:1689–1693
Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R (1996) Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev 59:3–10
Herter P, Kuhnen C, Muller KM, Wittinghofer A, Muller O (1999) Intracellular distribution of beta-catenin in colorectal adenomas, carcinomas and Peutz-Jeghers polyps. J Cancer Res Clin Oncol 125:297–304
Mahmoud NN, Boolbol SK, Dannenberg AJ, Mestre JR, Bilinski RT, Martucci C, et al (1998) The sulfide metabolite of sulindac prevents tumors and restores enterocyte apoptosis in a murine model of familial adenomatous polyposis. Carcinogenesis 19:87–91
Weiss H, Jacobasch KH, Haensch W, Streller B, Hieke B (1997) Significance of apoptosis in the process of tumorigenesis in colorectal mucosa and adenomas in FAP patients. Anal Cell Pathol 14:61–73
Peifer M (1997) Beta-catenin as oncogene: the smoking gun. Science 275:1752–1753
Muller-Decker K, Albert C, Lukanov T, Winde G, Marks F, Furstenberger G (1999) Cellular localization of cyclo-oxygenase isozymes in Crohn’s disease and colorectal cancer. Int J Colorectal Dis 14:212–218
Bamba H, Ota S, Kato A, Adachi A, Itoyama S, Matsuzaki F (1999) High expression of cyclooxygenase-2 in macrophages of human colonic adenoma. Int J Cancer 83:470–475
Acknowledgements
We thank Mrs. Ursula Becker for excellent technical assistance and Dr. Schäfer and Dr. Schulte-Witte for their support. This work was supported by the Ludwig-Demling-Stipendium (Olympus optical, Hamburg, Germany), the BONFOR-programme of the University of Bonn, Germany, and the Deutsche Krebshilfe (German cancer aid, Bonn, Germany).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jungck, M., Grünhage, F., Spengler, U. et al. E-cadherin expression is homogeneously reduced in adenoma from patients with familial adenomatous polyposis: an immunohistochemical study of E-cadherin, β-catenin and cyclooxygenase-2 expression. Int J Colorectal Dis 19, 438–445 (2004). https://doi.org/10.1007/s00384-003-0575-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00384-003-0575-z