
Abstract
Purpose
Variation in management characterizes treatment of infants with a congenital pulmonary airway malformation (CPAM). This review addresses six clinically applicable questions using available evidence to provide recommendations for the treatment of these patients.
Methods
Questions regarding the management of a pediatric patient with a CPAM were generated. English language articles published between 1960 and 2014 were compiled after searching Medline and OvidSP. The articles were divided by subject area and by the question asked, then reviewed and included if they specifically addressed the proposed question.
Results
1040 articles were identified on initial search. After screening abstracts per eligibility criteria, 130 articles were used to answer the proposed questions. Based on the available literature, resection of an asymptomatic CPAM is controversial, and when performed is usually completed within the first six months of life. Lobectomy remains the standard resection method for CPAM, and can be performed thoracoscopically or via thoracotomy. There is no consensus regarding a monitoring protocol for observing asymptomatic lesions, although at least one chest computerized tomogram (CT) should be performed postnatally for lesion characterization. An antenatally identified CPAM can be evaluated with MRI if fetal intervention is being considered, but is not required for the fetus with a lesion not at risk for hydrops. Prenatal consultation should be offered for infants with CPAM and encouraged for those infants in whom characteristics indicate risk of hydrops.
Conclusions
Very few articles provided definitive recommendations for care of the patient with a CPAM and none reported Level I or II evidence. Based on available information, CPAMs are usually resected early in life if at all. A prenatally diagnosed congenital lung lesion should be evaluated postnatally with CT, and prenatal counseling should be undertaken in patients at risk for hydrops.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The management of a patient with a CPAM is a controversial subject for pediatric surgeons. For example, a 2013 survey of the British Association of Pediatric Surgeons, to which 51% of the membership responded, highlighted the variability in the care of these patients [1]. While most respondents agreed that a postnatal chest X-ray and CT scan should be performed, half schedule the CT at 6 weeks of age while others obtain it between three and 12 months of age. Recommendations for resection also varied widely: 21% of respondents recommended mandatory resection, 24% recommended never resecting asymptomatic lesions, and 55% based the decision for resection on lesion size, parental anxiety, and desire for tissue diagnosis. Of the respondents who recommended resection, none performed resection prior to three months of age, 50.6% resected the lesion prior to 1 year of age, and 44% performed surgery after 1 year of age. The follow-up monitoring recommended by surgeons who advocated no surgical intervention for asymptomatic lesions also varied. Furthermore, while several recent reviews have evaluated literature surrounding CPAMs, none have done so on a truly systematic manner and they have focused on disparate topics related to either diagnostic and/or management options [2, 3].
Given the existing uncertainty surrounding best practices for CPAM, the purposes of this systematic review is to collate, evaluate, and synthesize the existing evidence for the management of CPAMs, and provide recommendations based on that evidence.
Materials and methods
Research questions
The American Pediatric Surgical Association (APSA) Outcomes and Evidence based practice (OEBP) committee selected the following questions for this systematic review:
-
1.
What is the long-term risk of an asymptomatic CPAM if observed?
-
2.
If observation is chosen, what is the observation strategy?
-
3.
What is the optimal operative approach (segmental/non-anatomic/lobar resection) for CPAM resection?
-
4.
What is the optimal timing for CPAM resection?
-
5.
What is the optimal imaging modality and timing of imaging for CPAM?
-
6.
What are the indications and outcomes for fetal intervention?
Search methods
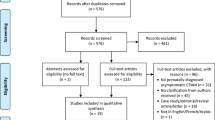
The search was performed with the aid of a health sciences librarian (see acknowledgement) who used broad Medical Subject Headings (MeSH) search terms that were inclusive and redundant to ensure completeness. The subject heading “cystic adenomatoid malformation of lung, congenital” was searched along with “congenital pulmonary airway malformation, CPAM, CCAM, congenital cystic airway malformation, and congenital cystic adenoid malformation” as keyword phrases. We followed Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) guidelines [4]. Using bibliography management software (EndNote, Thomson Reuters, New York, NY, USA), duplicates were removed, creating a master list of titles and abstracts. Two of the authors (C. C. and C. D.) independently scanned all abstracts. Articles were included or excluded based on their relevance to the topic and were narrowed by reading the abstract. Full-length papers were collected for further review. Animal studies, duplicates, non-English language publications, articles without an abstract, and general review articles were excluded (Fig. 1).

Diagram for screening and inclusion of abstracts and manuscripts for review
Data sources
Search terms were applied using the MEDLINE and OvidSP. Cochrane reviews were included in the MEDLINE search. Searches were limited to articles published from January 1, 1960 through January 31, 2014. In addition to the articles that were located through the search, the list was augmented by searching the reference list of each selected article to find other papers that may not have been included in the original search results. Articles published after the search date are included only to enhance recommendations but have not been subject to a formal review process.
Study selection
Studies selected for inclusion were critically appraised and ranked on their level of evidence based on the type of study (retrospective or prospective, randomized or not) per The Oxford Centre for Evidence Based Medicine (Table 1). A paucity of high-level evidence (level I and level II studies) required broadening the inclusion criteria to level III and level IV studies. For the purposes of reporting fetal intervention data, case reports were also included, given the relative lack of available reports regarding intervention modalities. However, opinion pieces, and editorials were excluded.
Data extraction
The Oxford Centre for Evidence Based Medicine criteria also guided the recommendations generated for each question. Data compiled for each article included sample size, description of the procedures performed, and outcomes including mortality and morbidity. A total of 1040 abstracts underwent initial title and abstract screening. Of these, 130 were selected for in-depth review of the full manuscript.
Results
What is the long-term risk of an asymptomatic CPAM if observed?
Will they become symptomatic?
Six articles addressed this question: one systematic review/meta-analysis, and five case series [5,6,7,8,9,10]. The systematic review provides the best overall analysis of this question, and three representative case series with the most detailed long-term follow-up and conflicting recommendations are discussed [5,6,7, 10].
A meta-analysis by Stanton and colleagues in 2009 evaluated whether elective or emergency surgery for CPAM was associated with a higher risk of adverse outcomes [5]. They compiled 41 reports that included 1070 patients, the majority of whom (79%) had antenatally detected lesions. Among these were 29 reports (871 cases) that described 505 (58%) asymptomatic antenatally detected patients who survived the perinatal period without surgery into infancy. Of these 505 initially asymptomatic patients, the CPAMs had a small (3.2%) risk of becoming symptomatic if they were not already symptomatic at birth. If patients developed symptoms, this usually occurred by ten months of age and included episodes of life-threatening sepsis and cardio-respiratory arrest. There was a twofold increase in operative risk if the lesion was resected after symptoms developed. These postoperative complications included air-leak, effusion, pneumonia, and bleeding; four patients died after emergency surgery for a previously asymptomatic lesion. Therefore, the authors recommend that elective resections should be performed prior to ten months of age.
In 2014 Ng et al. described 74 consecutive antenatally diagnosed CPAM patients over 10 years who were managed without initial surgery [6]. This included 72 live born infants, 5% of whom became symptomatic. One of these patients required an emergent operation as a neonate, two patients developed pneumonia and underwent lobectomy at 3 years of age, and one patient had a sequestration that was embolized at 5 years of age. Three asymptomatic patients underwent resection based on parental request at 8–12 months of age. Sixty-five patients remained asymptomatic and had not undergone surgical resection at a median follow-up of 5 years. Nine of these patients underwent further CT scans and 8/9 showed a decrease in size of the CPAM. The remaining 59 patients continue to follow-up clinically and no adverse consequences have been reported, though seven have had upper respiratory tract infections, four have symptoms of reactive airway disease, and the others remain symptom-free.
Khosa et al. reported that of 30 patients with a prenatal diagnosis of CPAM, nine developed symptoms within the first year of life [10]. Six of these patients were symptomatic at birth and underwent neonatal resection. Despite being asymptomatic at birth, three children in this series developed symptoms while awaiting surgery (infection at 7 and 11 months, and respiratory distress at 10 months).
Wong and colleagues from Children’s Hospital in Sydney, Australia published a series describing their care of 35 patients with CPAM from 1986 to 2007 [7]. Twenty-one patients were asymptomatic at birth. Eighteen (86%) of these initially asymptomatic patients became symptomatic at a median age of 2 years. Symptoms included pneumonia, respiratory distress, pneumothorax, and chronic cough. They concluded that the majority of patients who present with asymptomatic CPAM would subsequently become symptomatic and warrant referral and intervention to avoid development of complications.
In summary, many patients with CPAM are asymptomatic before or at birth. After the neonatal period, the likelihood of the lesion becoming symptomatic varies from 3 to 86% in reported series. Risk factors for predicting the development of symptoms could not be elucidated from available literature; additional information in future reports about lesion characteristics would aid in clarifying this issue. However, the general consensus across studies is that if one plans to operate, the procedure should be performed prior to the development of symptoms.
(Level IV evidence, Grade D recommendation)
Will they become malignant?
First described in 1988, pleuropulmonary blastoma (PPB) is a malignant lung lesion that shares many imaging characteristics with CPAM. Primarily due to cases of malignancy in specimens thought to be a benign CPAM prior to definitive resection, questions exist about the long-term risk of malignancy in unresected CPAM lesions. In a prior review article, Laberge identified reports of multiple types of malignancy, including sarcomas as well as bronchoalveolar carcinomas, in congenital lung lesions [11]. Two articles thought to best address this question, one pathology series and one case series, were reviewed and are here discussed [12, 13].
Priest and colleagues evaluated the risk of malignancy developing during early childhood in pulmonary cysts in a review based on the International PPB Registry, which included 220 pathologically confirmed cases of PPB [12]. Sixty-six percent of these cases were associated with lung cysts, and 29% were purely cystic lesions. In their report, PPB was not thought to be a malignant degeneration of a pre-existent CPAM, but was rather a completely different pathologic entity that could often not be distinguished radiographically from CPAM. Ninety-four percent of PPB lesions presented in children less than six years of age. Importantly, the report noted the difficulty in making a radiographic distinction between CPAM and PPB. In this series, several risk factors suggesting that a lesion was actually a pleuropulmonary blastoma and not a CPAM were presented. The authors state that operative resection of cystic lung lesions in children should be undertaken rather than observation if the lesions are associated with the development of a pneumothorax, bilateral lung cysts, multifocality, or a family history of similar lesions. Associated conditions that should also heighten concern for potential malignancy include renal cystic disease (including cystic nephroma), small bowel polyps, and additional childhood cancers or dysplasia.
Nasr and colleagues published a review of 129 CPAM and PPB patients in 2010, 74 of whom had undergone resection of the lesion [13]. Five of these patients had PPB on final pathology, three of whom had initially been thought to have CPAM rather than PPB. Pleuropulmonary blastoma, therefore, was present in 4% (3/74) of what were initially presumed to be benign lung lesions. No clinical or radiologic markers were found that could differentiate CPAM from PPB preoperatively and, therefore, resection of all CPAMs was recommended. This was a change to their recommendations from an earlier series which did not clearly support resection of asymptomatic lesions [14].
In summary, in radiographically identified CPAM lesions, there may be a 4% risk of pleuropulmonary blastoma, and an undefined risk for late malignant degeneration into other epithelial and mesenchymal malignancies such as bronchoalveolar carcinoma or rhabdomyosarcoma. It does not appear that pleuropulmonary blastoma arises directly from CPAMs, however, it may be difficult to distinguish between the two entities based on pre-operative imaging alone. A recently described algorithm based on clinical and radiographic features has been retrospectively described and may aid in surgical decision-making [15].
(Level IV evidence, Grade D recommendation)
If observation is chosen, what is the observation strategy?
Eighteen articles pertained to this question [1, 6, 7, 14, 16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Articles included two surveys and many case series. The surveys and the articles that best summarized specific follow-up recommendations are discussed, but all were utilized in forming our conclusions [1, 6, 19, 23, 28, 29]. There was no level I, II, or III evidence available.
Variability in postnatal follow-up of patients with asymptomatic CPAMs seems to be the rule rather than the exception. In 2006, Lo and colleagues surveyed members of the Canadian Association of Paediatric Surgeons (CAPS) regarding their recommendations for follow-up of CPAM patients who were asymptomatic at birth [19]. Forty-nine surgeons responded (69% of the membership). This survey demonstrated high inter- and intra-institutional variability regarding early resection, late resection, or observation of CPAM patients. There was also significant variation in the type and frequency of radiographic studies, including chest X-ray, ultrasound, CT scan, magnetic resonance imaging (MRI), and a combined modality evaluation. The duration of follow-up was also highly variable. They concluded that the management approach of individual pediatric surgeons was based on personal interpretation of the available limited literature. These findings were confirmed by Peters and colleagues with their similar survey of the British Association of Pediatric Surgeons in 2013 which yielded very similar results [1].
Van Leeuwen and colleagues at the University of Michigan identified 14 patients over 10 years who were diagnosed prenatally with a CPAM [28]. Four of these patients were symptomatic at birth and underwent resection as neonates. The remaining ten patients underwent CT scan soon after birth, all of which identified the prenatally diagnosed lung lesion. Three of these ten patients underwent lung resection at 2 months, 9 months, and 5 years of age based on surgeon preference. They felt that there was adherence to an initial management plan in these patients, but found that the decision to resect a CPAM in an asymptomatic patient was ultimately based on the preferences of each individual pediatric surgeon.
In 2008, Tran and colleagues reported on 38 prenatally diagnosed CPAM patients [29]. Four pregnancies were complicated by hydrops fetalis, and two of these hydropic infants died (one in utero and one as a neonate). Ten patients diagnosed with CPAM in utero appeared to have complete resolution of the CPAM on antenatal ultrasound, but six had an abnormal postnatal chest X-ray, and eight of these ten infants had CT scans, all of which were abnormal. In all, 23 of the 33 babies who survived and were asymptomatic after the newborn period underwent CT, and all were abnormal. The timing of CT scan ranged from two days of age to four years of age. Twenty-seven percent of the patients had undergone resection at the time of the report.
Sauvat and colleagues reported on 29 patients antenatally diagnosed with CPAM who were asymptomatic at birth over a 10-year time period [23]. This is a subset of a total of 75 patients referred to their institution with CPAM over the same time period. In the asymptomatic patients, the CPAM “vanished” in six patients, meaning that the lesion was not visible on both chest X-ray and CT scan. CT was recommended for the postnatal evaluation of CPAM, but could be postponed until day of life 45 if the patient was asymptomatic. If the CT, obtained either soon after birth or at 45 days of age, showed a 3 cm or larger lesion or fluid within the lesion, they recommended surgical resection, otherwise a follow-up CT 6–12 months later was recommended. As a result of these criteria, 17 patients underwent resection in the first months of life, and the remaining 12 patients are being followed.
Ng et al. published a 5-year follow-up to their prior report on 65 initially asymptomatic patients managed by observation [6]. They classified patients as appropriate for surveillance if they had antenatally diagnosed lesions that were unilateral, in a single lobe, and had no suspected genetic predisposition to malignancy. They performed an initial CT scan at 3–6 months of age and did not repeat imaging in the absence of symptoms. Five patients had no visible cystic lung lesion on CT and were not followed further. Two patients developed pneumonia and underwent resection at 3 years of age, and another required embolization of a sequestration at 5 years of age. The remaining 57 are followed clinically, with nine undergoing further CT scans, most of which (8/9) showed a reduction in size of the lung lesion.
In summary, observation strategies for asymptomatic CPAMs generally include at least one postnatal CT scan. The timing of the CT scan depends on the practitioner’s preferred management strategy and potential timing of resection. Patients may be evaluated differently if they develop symptoms such as pneumonia or pneumothorax. Surgical resection should be advocated if a patient has a family history that may predispose them to pleuropulmonary blastoma or there is concern for DICER1 syndrome [30].
(Level IV evidence, Grade D recommendation)
What is the optimal operative approach (segmental/non-anatomic/lobar resection) for CPAM resection?
Lobectomy has generally been considered the most common surgical treatment of a patient with a CPAM confined to a single lobe. This section will address the question of CPAM resection via an approach other than lobectomy such as: segmentectomy, “wedge” resection or another non-anatomic resection. For the purposes of this paper, non-lobectomy resections will be termed lung-sparing resections (LSR). Given that lobectomy has long been considered the standard of care in pediatric surgery for a CPAM occupying one lobe, the issue addressed here is whether LSR for the treatment of a patient with an asymptomatic, single lobe lesion is inferior, equivalent, or superior to lobectomy.
Twenty-nine studies addressed the question of surgical approach, and nine manuscripts included patients who underwent LSR (Table 2). In these nine reports of 127 patients, LSR was performed using either an open or thoracoscopic approach, and variable lengths of postoperative follow-up were reported. Although no patient experienced a malignancy during follow-up, it is difficult to draw any definitive conclusions from this observation as malignancy tends to present in the second or third decade of life and long-term follow-up was not consistently reported. In addition, although the operations were performed safely (no operative mortality), 11 patients (9%) required reoperation for persistent air leak, bronchopleural fistula, or persistent disease detected on follow-up imaging, which was itself not performed routinely in all studies.
In 1981, Nishibayshi described five patients who underwent LSR for symptomatic CPAM [31]. Four of the patients experienced significant postoperative sequelae such as empyema (one patient), prolonged air leak (two patients), or residual disease requiring reoperation (one patient). Conversely, Browdie and co-authors reported three patients with symptomatic multifocal disease who underwent LSR and had uneventful recoveries apart from a longer period of air leak from the chest tube (5–7 days) [32]. Follow-up ranged from 5 to 19 years and none of the three patients had clinical complications. Sapin et al. identified six patients from a prenatally diagnosed cohort who eventually underwent LSR; one underwent completion lobectomy at a later time for concerning findings on follow-up imaging [33]. Waszak and others from Lyon, France reported ten patients (seven asymptomatic) who underwent segmentectomy for CPAM, of whom three had a prolonged air-leak and one required re-operation for residual disease [34]. It is worth acknowledging that modern technical tools for tissue sealing (reinforced staple loads, fibrin sealants, radiofrequency energy sealing devices) were not available at the time of that study.
A study from Seoul, South Korea reported on 45 consecutive patients who underwent surgical resection for CPAM over a 12-year period [35]. This was a heterogeneous cohort, with the majority of patients undergoing resection at a later age (mean age at operation 11.4 years) for symptomatic disease (64%). Thirty-three patients were treated by lobectomy and 12 underwent LSR. Prolonged air leak occurred in one patient in each group and the average length of stay was not different between groups. In long-term follow-up of the LSR group, one patient had an asymptomatic residual lesion on a follow-up CT scan but did not undergo repeat resection. No mention was made with regard to long-term pulmonary function.
Johnson et al. reported their experience with thoracoscopic LSR in 15 patients between 2007 and 2010 [36]. Of the patients, 12 (80%) were diagnosed prenatally and nine were asymptomatic at the time of resection. Parenchymal resection was carried out by direct visualization of the lesion and definition of “radiographic borders” using either a vessel sealing radiofrequency energy device or a GIA stapler. The mean length of stay was 4.2 days. During follow-up, all patients underwent a CT scan six weeks postoperatively; one patient required reoperation for what was thought to be an incomplete initial resection.
The systematic review by Stanton summarized the surgical approach when available [5]. The type of surgery undertaken was specified in 328 cases, of which 268 were lobectomy and 60 were segmentectomy. Residual disease was reported as a complication in nine patients (15%) who had undergone segmental resection (age at surgery not specified in all) while no patients had reported residual disease after lobectomy. The largest reported series of LSR for CPAM (54 patients over 10 years) comes from the University of Bologna, Italy [37]. Patients were judged to be suitable for LSR if the lesion was surrounded by normal lobar parenchyma on CT and identified visually at the time of surgery. Twenty-six were approached thoracoscopically, with 18 cases requiring conversion to thoracotomy. There were six postoperative complications in the 54 patients: three asymptomatic pneumothoraces that resolved without intervention, one tension pneumothorax that required replacement of thoracostomy tube, and three instances of intra-operative bleeding requiring blood transfusion. The mean duration of follow-up was 65 months. All patients underwent a chest radiograph 1 year after surgery and a subsequent chest CT if the chest radiograph or symptoms raised the suspicion of residual disease. Two patients experienced pneumonia during the follow-up period. A third patient had a cystic lung lesion on postoperative CT that required lobectomy. The authors concluded that LSR is a safe and effective means of surgical treatment for carefully selected patients with CPAM—namely, asymptomatic patients with small lesions identified with a high-quality CT scan.
Bagrodia et al. recently published another series of patients undergoing segmentectomy for CPAM [38]. Thirty-four patients had an intrapulmonary lesion (CCAM or intrapulmonary sequestration) resected by either lobectomy (n = 15) or LSR (n = 19). Patients undergoing LSR had a shorter length of stay than their lobectomy counterparts and a slightly shorter duration of chest tube dwell. However, 80% of the patients undergoing LSR were approached thoracoscopically, whereas the majority of the patients undergoing lobectomy were approached through a thoracotomy. Although operative morbidity was low, they reported no long-term follow-up and did not study postoperative pulmonary function after either lobectomy or LSR. Sauvat and co-authors reported 11 cases of LSR but did so in the context of a paper addressing observation for prenatally diagnosed lesions; no mention was made of specific short and long-term outcomes of LSR compared to lobectomy [23].
In summary, lobectomy remains the standard of care for surgical resection for most patients with CPAM. Whereas LSR is feasible and safe, long-term follow-up after LSR is inconsistent, and there is no information regarding the benefit of LSR on long-term pulmonary function. There is no current evidence that LSR is superior to lobectomy for a single lobar asymptomatic CPAM either in terms of perioperative morbidity or long-term pulmonary function.
(Level IV evidence, Grade D recommendation)
What is the optimal timing for an asymptomatic CPAM resection?
Wide variation exists among pediatric surgeons regarding the optimal timing of resection of an asymptomatic CPAM. To answer the question of the optimal window for resection, we reviewed twelve studies that specifically addressed timing of surgery. Due to a paucity of high-level data, the following analysis also draws on other literature to comment on the rationale for timing resection of CPAM.
What is the ideal time to provide for optimal compensatory lung growth after pulmonary resection?
A large body of animal data supports the concept that compensatory lung growth occurs after lobectomy or pneumonectomy; however, the nature and extent of that growth is not clear. Studies evaluating compensatory growth after pulmonary resection in animals of varying ages are not entirely consistent. Although the rate of compensatory growth is likely faster in younger animals, the quantitative compensatory response may not be significantly different from that of older animals. Taken together, these observations suggest that the mechanisms for compensatory lung growth are likely different from those of normal lung growth, but compensatory development does indeed occur [39].
In 1998, Nakajima and colleagues reported the long-term pulmonary function of 27 patients who had undergone lobectomy in infancy or childhood [40]. Although vital capacity (%VC) decreased immediately after lobectomy, it recovered to normal values within two postoperative years and remained within or above the normal range. However, in patients who underwent lobectomy after 4 years of age, %VC was significantly lower compared to patients who underwent resection at an earlier age.
In 2009, Komori and co-authors examined long-term pulmonary function in 93 patients who underwent lobectomy between 2 days to 15 years of life [41]. Alveolar growth (as opposed to emphysematous change) as measured by radionucleotide imaging was lower in patients who had undergone lobectomy after 1 year of age, suggesting an improvement in long-term pulmonary function in those patients who had undergone lobectomy prior to 1 year of age. In addition, those patients who had a history of preoperative pulmonary infection had more emphysematous lung growth after lobectomy regardless of the age at resection, suggesting that an operation prior to the development of an inflammatory process would result in more effective alveolar growth as opposed to emphysematous compensation.
Conversely, Keijzer et al. examined a small cohort of 14 patients who underwent traditional pulmonary function testing at the age of 10 years after undergoing lobectomy at an earlier age and found no difference in mean forced vital capacity (FVC) or mean forced expiratory volume in 1 s (FEV1) between patients undergoing lobectomy before the age of 2 years compared with those having the operation after 2 years of age [42]. A similar study by Naito et al. also essentially demonstrated normal PFTs after lobectomy in 28 patients whose operation occurred between 3 days and 56 months of age [43]. There was a trend towards inferior VO2max measurements in older patients but this failed to reach significance (p = 0.055). Both studies were limited by very small sample sizes.
In summary, the literature provides inconsistent results regarding pulmonary function in humans after pulmonary resection. Nevertheless, current evidence supports the practice of early resection based on the possibility of compensatory lung growth. The literature, however, does not clearly identify an age at which the intersection between operative risk and compensatory lung growth occurs.
(Level IV evidence, Grade D recommendation)
What is the ideal time to resect a CPAM to prevent the development of inflammation or infectious complications?
The principal aim in offering resection to a patient with an asymptomatic CPAM is to prevent symptoms or complications, most commonly pneumonia or lung abscess. Performing a lobectomy prior to the development of inflammatory changes in the chest is also preferable as it may theoretically make the operation easier and decrease operative time and perioperative complications.
Calvert and coauthors reported their experience with 13 patients with CPAM who were asymptomatic and underwent resection at ages either less than or greater than 6 months of age [44]. There were histologic or gross inflammatory changes in the resected specimens in 50% of patients who underwent surgery after 6 months, compared to only 20% in those who had a resection prior to 6 months of age. Pelizzo et al. also demonstrated histologic findings of inflammation in 50% of lobectomy specimens taken from patients undergoing elective lobectomy for an asymptomatic CPAM at 3 months of age [45]. Neither of these studies commented on operative difficulty or incidence of perioperative complications associated with these histologic changes.
Conforti and colleagues reported a cohort of 57 consecutive patients with a CPAM over an 8-year period [46]. Among the 35 children who were asymptomatic at birth and later underwent lobectomy, there was no difference in length of hospital stay or morbidity between those patients who underwent resection prior to 6 months of age compared to after 6 months of age. However, there was an increased incidence of developing respiratory symptoms before surgery in patients undergoing surgical resection after 6 months of age. In addition, patients who had symptoms prior to surgery experienced a more complicated (longer hospital stay, more ventilator days, longer time of pleural drainage) postoperative clinical course compared with asymptomatic patients.
Kim and co-authors described another single-institution experience of 40 patients over a 6-year period [47]. They found that older age at the time of surgery was associated with the higher risk of developing operative complications, although all patients eventually recovered. The odds ratio of developing an operative complication per month of increased age was 1.025 (CI 1.002–1.048). These findings suggested that a “delay of months” would not pose a significant perioperative risk of morbidity, but waiting several years may result in substantial perioperative risk. The authors did not specify the number of months that would be safe to wait. They performed four elective resections in patients with prenatally diagnosed CPAM within the first month of life and those patients had no increased operative morbidity when compared to their older counterparts.
Marshall et al. retrospectively described 16 initially asymptomatic patients who eventually underwent lobectomy [48]. Surgical resection in the asymptomatic period for those patients who were diagnosed prenatally was associated with a shorter length of stay and a trend toward a decreased major postoperative complication rate compared with patients who did not have a prenatal diagnosis and presented with symptoms (respiratory distress, infection) at a later date.
At the Children’s Hospital of Philadelphia over a 10-year period, 170 patients received postnatal care for a prenatally diagnosed fetal lung lesion [49]. One hundred nine patients were asymptomatic at birth. Four of these patients underwent a thoracoscopic lobectomy and were excluded from further analysis. The remaining patients (n = 105) underwent chest CT to confirm the prenatal diagnosis and then had surgical resection by open thoracotomy at a mean age of 2.5 months. Of these 105 patients, seven developed a complication that included a prolonged postoperative air leak in three patients and need for a transfusion in four patients—two for intraoperative bleeding and two for preoperative anemia. The average length of stay was 3 days, and there was no perioperative mortality.
Rothenberg et al. reported their experience with 74 patients weighing less than 10 kg who successfully underwent thoracoscopic lobectomy [50]. Twenty-six of these patients underwent lobectomy at less than 3 months of age prior to the development of symptoms. Their mean length of hospital stay was 1.5 days, and there were no perioperative complications noted. The practice of performing thoracoscopic lobectomy in pediatric patients was first advocated by Albanese and co-authors who safely performed 14 thoracoscopic lobectomies on small children (mean age 6 months) using a vessel-sealing device for vascular dissection [51]. Both Rothenberg and Albanese editorialized that by operating early on younger patients, the inflammatory changes associated with both clinically apparent and subclinical infections may be avoided. Although they and others have suggested that minimally invasive lobectomy can be more challenging once the child has already suffered a clinical chest infection there is no objective data to support this claim [52, 53].
In summary, with regard to the “optimal timing” for resection, immediate postnatal resection is not mandatory for the asymptomatic patient. While there is little evidence to mandate an operation at a defined time in the first year of life, the current available data would suggest that early (<6 months of age) surgery allows for ease of operative intervention, adequate recovery, and a reasonable time for compensatory lung growth while avoiding the potential infectious complications associated with observation.
(Level IV evidence, Grade D recommendation)
What is the optimal imaging modality and timing of imaging for an asymptomatic CPAM?
We reviewed 31 studies with a focus on prenatal and postnatal imaging, with only 11 of the studies addressing postnatal imaging modalities. No studies compared postnatal axial imaging techniques in terms of timing, accuracy, and efficacy.
Prenatal imaging
A pulmonary parenchymal abnormality noted on a prenatal screening ultrasound often prompts referral to a maternal-fetal medicine specialist for a confirmatory ultrasound examination. In addition to characterization of the lesion, an assessment for other anomalies and hydrops, a measurement of CVR (CPAM volume ratio) is often performed. The CVR is calculated by taking the product of the three dimensional measurements of the lesion (height × width × length) and 0.523 (the formula for the volume of an ellipse) and dividing this result by the head circumference. Crombleholme and colleagues first described the CVR and found that a measurement greater than 1.6 at the initial fetal ultrasound investigation predicted an increased risk of hydrops developing in a patient with a CPAM [54]. They concluded that a CVR ≤1.6 at presentation suggests that the risk of hydrops developing in the absence of a dominant cyst is less than 3%. Yong and co-authors reported a retrospective cohort of 71 consecutive cases of prenatally diagnosed CPAM and confirmed that a CVR >1.6 was significantly associated with hydrops [55]. In addition, a higher CVR correlated with adverse postnatal outcome at a mean age of follow-up of 41 months (range <1 to 117 months) and noted that a CVR <0.56 was predictive of an asymptomatic patient at birth. In a another retrospective review of CVR, Cass and co-authors reviewed 82 consecutive fetuses evaluated for a lung mass at a single fetal treatment center CVR correlated strongly with the development of hydrops and the need for fetal therapy [56]. This group noted that the CVR threshold for need for fetal intervention was more than two.
There is little data to guide what ultrasound characteristics should prompt fetal MRI. Compared to high quality ultrasound, routine MRI often does not add clinically relevant prenatal information when the diagnosis is clear [57, 58]. However, Hubbard et al. described an initial experience with fetal MRI at a single fetal treatment center in 18 patients referred from outside hospitals based on an ultrasound diagnosis and concluded that the fetal MRI changed the prenatal diagnosis in nine fetuses (50%) [59]. In that study, the ultrasound results were not reviewed and if an ultrasound was repeated at the fetal treatment center those results were not elucidated in the manuscript. All nine patients had an ultrasound diagnosis of a CPAM at referral—three patients were found to have a CDH, one tracheal atresia, one pulmonary agenesis, one neuro-enteric cyst, one bronchial stenosis, and one with bronchopulmonary sequestration. The other patient was thought to have bilateral CPAM on ultrasound, but MRI confirmed a large unilateral CPAM. Alternatively, Matsuoka described seven patients who underwent fetal MRI in the setting of an ultrasound diagnosis of a fetal thoracic lesion that they felt to be “atypical or complicated by other anomalies” [60]. Six of the seven patients had the diagnosis confirmed, but not changed by the fetal MRI, and the only diagnosis that was altered by MRI was a fetus that was noted to have a pleural effusion and polyhydramnios on ultrasound, but was found to have esophageal atresia in addition to those findings and died soon after cesarean delivery at 30 weeks due to worsening fetal hydrops. Liu and colleagues retrospectively evaluated 20 cases where ultrasound suggested a complex fetal cystic lung lesions to evaluate the effectiveness of MRI [61]. Of ten patients who were though to have a CPAM based on ultrasound, five were confirmed to have a CPAM on MRI whereas fetal MRI demonstrated a bronchopulmonary sequestration (BPS) in three, and congenital lobar hyperinflation in two. In ten patients where the ultrasound suggested a BPS, eight were found to have a BPS on MRI and the other two a CPAM. Although fetal MRI was a complementary tool in differentiating among BPS, CPAM, and congenital lobar fluid overload, the authors did not discuss how the MRI affected prenatal or postnatal management apart from the benefits of more accurate prenatal counseling.
Fetal MRI may alter the prenatal diagnosis in some patients who are initially felt to have a CPAM on fetal ultrasound. Clearly, an accurate diagnosis is mandatory prior to fetal intervention, but the benefits are less clear when considering prenatal counseling—especially as it relates to differentiating between a CPAM and BPS, where postnatal management is dependent on postnatal imaging regardless of the prenatal findings. To date, the number of patients for whom an MRI truly adds or changes a well-performed ultrasound remains poorly described outside of large prenatal referral centers. The limited studies included in this review did not address the expertise of the fetal ultrasonographer or the benefit of repeat ultrasonography when the initial diagnosis may be in question or performed in a screening capacity.
Postnatal imaging
For the asymptomatic patient with a prenatally diagnosed lesion, a single anterior-posterior radiograph is generally obtained as an initial method to identify issues that may require immediate intervention. However, there is no objective information to suggest how often this radiograph changes management in the asymptomatic infant. It is clear that a normal postnatal chest radiograph is not a reliable indicator of resolution of a lesion noted on prenatal ultrasound [62]. Definitive imaging to confirm a suspected CPAM on prenatal imaging in the asymptomatic patient is often carried out by multidetector computerized tomography with angiography (MDCTA). This technique allows for rapid acquisition of high-resolution 3-D datasets and reduces the need for sedation, which is advantageous for the neonate and infant [63]. Magnetic resonance imaging (MRI) provides an alternative to CT for the evaluation of an asymptomatic CPAM; however, as a first-line modality for the evaluation of congenital lung anomalies, MR is limited given the suboptimal capability of MR imaging to assess lung parenchymal abnormalities accurately. We found no literature that described a meaningful experience with postnatal MRI in the patient with an asymptomatic CPAM.
In summary, prenatal fetal lung lesions are commonly followed by fetal ultrasound, and fetal MRI should be considered in high-risk lesions when fetal therapy is being entertained or when the diagnosis is unclear. There is little data to suggest that routine MRI is warranted in all cases of prenatal cystic lung anomalies. Plain radiography immediately after birth typically adds little value for the asymptomatic patient, but may detect findings that may prompt earlier resection. Postnatal multidetector chest CT angiography (MDCTA) should be performed in patients in whom any lesion has been noted prenatally to confirm the presence of the lesion and characterize its location and potential extra-pulmonary blood supply. The timing of this study is variable.
(Level V evidence, Grade D recommendation)
What are the indications and outcome for fetal intervention?
Question 6 addresses the fetus with an in utero diagnosis of a cystic lung lesions thought to be a congenital pulmonary airway malformation. Thirty-six studies were further reviewed; we discarded singular case reports, but retained literature summaries of existing individual reports. We did not encounter any level I, II, or III evidence to assist with answering this question. Particular attention was paid to prenatal indications for intervention and/or treatment.
A fetus with a cystic lung lesion is determined to be “high risk” based on imaging characteristics either by ultrasound, echocardiography, or MRI. Prenatal characteristics of high risk include a CVR >1.6, placentomegaly, abnormal fetal echocardiography, diaphragm eversion, severe mediastinal shift, or lung hypoplasia (on MRI). Maternal mirror syndrome is the constellation of fetal and placental hydrops with maternal preeclampsia and portends a poor prognosis for the fetus. All of these conditions can occur in the fetus with a cystic lung lesion, and when they do, merit consultation with a fetal treatment center (see Tables 3, 4).
Prenatal steroid therapy
Tsao et al. from the University of California, San Francisco (UCSF), first elucidated the effectiveness of maternal steroids for the treatment of a high-risk fetus with a congenital cystic lung lesion [51]. Three mothers who were referred to UCSF for fetal surgical resection of a microcystic lung lesion were given preoperative betamethasone as routine medication prior to fetal surgery. In these three cases, fetal surgery was ultimately abandoned, the mild/moderate hydrops resolved, and the pregnancy continued to term. The three infants did not require an immediate postnatal resection. Since that first description, others have reported a similar experience in patients with microcystic lesions that is summarized in Table 5 [64,65,66,67]. Maternal betamethasone is given as two doses of 12 mg intramuscularly with 24-h between doses. The “variable response to steroids” as noted by Morris and colleagues was variable in the sense that all of the patients who were “non-responders” had macrocystic lesions [67]. Of the 11 patients who were “responders”, one had a favorable initial response, eventually underwent fetal lobectomy at 23 weeks, and suffered in utero fetal demise 5 weeks later. Two of the patients had macrocystic disease but, in addition to steroids, were treated with a thoracoamniotic shunt. The remainder of the responders had microcystic disease and survived to delivery and beyond. No adverse maternal or fetal events have been noted resulting from betamethasone administration.
Prenatal cyst decompression
Trans-amniotic needle decompression or thoracoamniotic shunting for patients with macrocystic lesions has been described in 28 case reports that were not part of the final 130 papers thoroughly reviewed. In a systematic review of in utero pulmonary drainage in the management of a congenital cystic lung lesion by Knox et al., a sub-analysis of studies that compared drainage (needle decompression or shunting) with non-treatment in fetuses complicated by hydrops revealed a marked association of drainage with survival [68]. The authors noted that there is a paucity of good quality evidence to inform clinical practice reliably regarding intrauterine pulmonary drainage for CPAM, but the available evidence suggests that perinatal survival may be improved by in utero treatment for those cases with macrocystic congenital cystic lung lesions and hydrops. Nevertheless, no single study directly compares tapping to shunting, thus we cannot directly comment on the merits of one versus the other.
Timing and fetal surgery
For the fetus that does not respond appropriately to a non-operative fetal intervention and is less than 32 weeks of gestational age, fetal lobectomy may be considered. Harrison and the fetal treatment group from UCSF described the first successful fetal lobectomy in 1990 [69]. In 1998, Adzick updated the cumulative experience with fetal lobectomy performed between 21 and 29 weeks gestation in 13 cases with eight survivors, but noted that all of the patients who underwent fetal lobectomy had developed hydrops and most assuredly would have died had lobectomy not been performed [70]. In the eight fetuses that survived, CPAM resection led to hydrops resolution in 1–2 weeks, return of the mediastinum to the midline within 3 weeks, and noticeable in utero lung growth. Developmental testing every 6–12 months had been normal in all eight survivors at the time of their report. Today, fetal lobectomy is offered less frequently due to the excellent response of many microcystic lesions to maternal steroid therapy and the heightened understanding of the importance of serial ultrasound examination and close follow-up for patients with a known CPAM. However, it remains a feasible option in fetal centers for those who fail to respond to other therapies.
Some fetal centers have had a favorable experience in selected cases with a controlled operative (Caesarean) delivery with fetal lobectomy while on placental support—the ex-utero intrapartum treatment (EXIT) to resection strategy. A fetal lobectomy is carried out on placental support when there is persistent mediastinal shift and/or hydrops associated with a persistently elevated CVR after 32 weeks of gestational age. Hedrick and colleagues published an initial experience of nine patients undergoing EXIT to resection of whom eight survived [71]. The maternal morbidity at the time of the report was limited to preterm labor (four patients) and chorioamnionitis (one patient). That same group anecdotally reported a subsequent experience of 22 patients with 20 survivors and similar maternal morbidity in response to a presentation from Cass and colleagues who also reported a similar successful experience with EXIT to resection in their center [72]. Cass and colleagues described opening the fetal chest while on placental support, but proceeding with removal from placental support if the fetus could be adequately ventilated. If adequate ventilation was achieved, the fetus was taken to a separate operating room and the lobectomy was completed while a separate surgical team cared for the mother. All nine patients who underwent EXIT to resection for persistent mediastinal compression from a CPAM in this series survived to discharge. Prior to implementing this strategy, four of six fetuses with significant mediastinal compression died using a strategy of conventional resection after delivery.
In summary, in the absence of randomized studies and long-term follow-up, identification of the fetus that could clearly benefit from in utero or ex-utero intrapartum treatment to improve perinatal and postnatal outcome is unclear. For patients with a CPAM and developing hydrops, consultation with a fetal treatment center likely offers benefit for the fetus and family. While there are no data to support this recommendation, fetal treatment modalities are not widely available and there are data to support the case for fetal treatment. Examination of the data allows for these recommendations:
-
Prenatal steroids should be administered to mothers carrying a fetus with a microcystic congenital cystic lung lesion with high risk factors.
(Level IV evidence, Grade C recommendation)
-
Thoracoamniotic shunting or decompression should be offered when a fetus has a macrocystic congenital cystic lung lesion with “high risk” factors and hydrops.
(Level IV evidence, Grade D recommendation)
Summary and conclusions
This review elucidates the paucity of data to allow for clear recommendations for the management of the fetus, neonate, and child with an asymptomatic CPAM. There is clearly a need for risk-adjusted, lesion stratified data collection that is likely best performed at a multi-institutional level. After the neonatal period, the likelihood of an asymptomatic lesion becoming symptomatic is upwards of 85%; yet reproducible risk factors for predicting the development of symptoms are not currently available. Variability in postnatal follow-up of patients with asymptomatic CPAMs is the rule rather than the exception, and the optimal timing of surgery is not well known. The available evidence would support a variety of approaches based on conflicting and low level evidence, but if surgical resection is chosen, doing so prior to the development of symptoms is justifiable based on the available evidence. We do know of current ongoing trials evaluating the promise of observation of selected asymptomatic lesions, and the results of that work will certainly add to our knowledge of whether or not it is safe to observe the child with an asymptomatic CPAM. Ongoing assessment of long-term pulmonary function in patients undergoing operative intervention is necessary to evaluate the merits of lung sparing interventions vs. standard lobectomy as well as the appropriate timing of surgical extirpation of an asymptomatic lesion. The utility of prenatal MRI remains largely unknown in cases of an uncomplicated chest lesion noted on routine prenatal ultrasonography, but does appear to assist in the differentiation of lesions when the ultrasound diagnosis is unclear or a fetal intervention is planned. An optimal postnatal imaging protocol cannot be recommended from the literature reviewed, but axial imaging does clearly offer a level of anatomic detail that cannot be elucidated by thoracic ultrasound or plain radiography. For the fetus with high risk factors for developing hydrops, prenatal steroids should be administered to mothers carrying a fetus with a microcystic CPAM and thoracoamniotic shunting is a useful fetal interventional modality when faced with a cystic space occupying thoracic lesion.
References
Peters RT, Burge DM, Marven SS (2013) Congenital lung malformations: an ongoing controversy. Ann R Coll Surg Engl 95(2):144–147
Baird R, Puligandla PS, Laberge JM (2014) Congenital lung malformations: informing best practice. Semin Pediatr Surg 23(5):270–277. doi:10.1053/j.sempedsurg.2014.09.007
Kapralik J, Wayne C, Chan E, Nasr A (2016) Surgical versus conservative management of congenital pulmonary airway malformation in children: a systematic review and meta-analysis. J Pediatr Surg 51(3):508–512. doi:10.1016/j.jpedsurg.2015.11.022
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, Clarke M, Devereaux PJ, Kleijnen J, Moher D (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med 6(7):e1000100. doi:10.1371/journal.pmed.1000100
Stanton M, Njere I, Ade-Ajayi N, Patel S, Davenport M (2009) Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg 44(5):1027–1033
Ng C, Stanwell J, Burge DM, Stanton MP (2014) Conservative management of antenatally diagnosed cystic lung malformations. Arch Dis Child 99(5):432–437
Wong A, Vieten D, Singh S, Harvey JG, Holland AJ (2009) Long-term outcome of asymptomatic patients with congenital cystic adenomatoid malformation. Pediatr Surg Int 25(6):479–485
Wang A, D’Amico TA, Berry MF (2014) Surgical management of congenital pulmonary malformations after the first decade of life. Ann Thorac Surg 97(6):1933–1938
Kamata S, Usui N, Kamiyama M, Nose K, Sawai T, Fukuzawa M (2006) Long-term outcome in patients with prenatally diagnosed cystic lung disease: special reference to ventilation and perfusion scan in the affected lung. J Pediatr Surg 41(12):2023–2027
Khosa JK, Leong SL, Borzi PA (2004) Congenital cystic adenomatoid malformation of the lung: indications and timing of surgery. Pediatr Surg Int 20(7):505–508
Laberge JM, Puligandla P, Flageole H (2005) Asymptomatic congenital lung malformations. Semin Pediatr Surg 14(1):16–33
Priest JR, Williams GM, Hill DA, Dehner LP, Jaffe A (2009) Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol 44(1):14–30. doi:10.1002/ppul.20917
Nasr A, Himidan S, Pastor AC, Taylor G, Kim PC (2010) Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg 45(6):1086–1089
Aziz D, Langer JC, Tuuha SE, Ryan G, Ein SH, Kim PC (2004) Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation: to resect or not? J Pediatr Surg 39(3):329–334 (discussion 329–334)
Feinberg A, Hall NJ, Williams GM, Schultz KA, Miniati D, Hill DA, Dehner LP, Messinger YH, Langer JC (2016) Can congenital pulmonary airway malformation be distinguished from Type I pleuropulmonary blastoma based on clinical and radiological features? J Pediatr Surg 51(1):33–37. doi:10.1016/j.jpedsurg.2015.10.019
Hammond PJ, Devdas JM, Ray B, Ward-Platt M, Barrett AM, McKean M (2010) The outcome of expectant management of congenital cystic adenomatoid malformations (CCAM) of the lung. Eur J Pediatr Surg 20(3):145–149
Hsieh CC, Chao AS, Chang YL, Kuo DM, Hsieh TT, Hung HT (2005) Outcome of congenital cystic adenomatoid malformation of the lung after antenatal diagnosis. Int J Gynaecol Obstet 89(2):99–102
Kongstad T, Buchvald F, Brenoe J, Petersen BL, Tabor A, Nielsen KG (2012) Radiology, histology and short-term outcome of asymptomatic congenital thoracic malformations. Acta Paediatr 101(2):155–158
Lo AY, Jones S (2008) Lack of consensus among Canadian pediatric surgeons regarding the management of congenital cystic adenomatoid malformation of the lung. J Pediatr Surg 43(5):797–799
Mentzer SJ, Filler RM, Phillips J (1992) Limited pulmonary resections for congenital cystic adenomatoid malformation of the lung. J Pediatr Surg 27(11):1410–1413
Revillon Y, Jan D, Plattner V, Sonigo P, Dommergues M, Mandelbrot L, Dumez Y, Nihoul-Fekete C (1993) Congenital cystic adenomatoid malformation of the lung: prenatal management and prognosis. J Pediatr Surg 28(8):1009–1011
Roggin KK, Breuer CK, Carr SR, Hansen K, Kurkchubasche AG, Wesselhoeft CW Jr, Tracy TF Jr, Luks FI (2000) The unpredictable character of congenital cystic lung lesions. J Pediatr Surg 35(5):801–805
Sauvat F, Michel JL, Benachi A, Emond S, Revillon Y (2003) Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg 38(4):548–552
Sueyoshi R, Okazaki T, Urushihara N, Fujiwara T, Tobayama S, Fukumoto K, Horigome F, Tei E, Lane GJ, Hasegawa S, Yamataka A (2008) Managing prenatally diagnosed asymptomatic congenital cystic adenomatoid malformation. Pediatr Surg Int 24(10):1111–1115
Takeda S, Miyoshi S, Inoue M, Omori K, Okumura M, Yoon HE, Minami M, Matsuda H (1999) Clinical spectrum of congenital cystic disease of the lung in children. Eur J Cardiothorac Surg 15(1):11–17
Abecasis F, Gomes Ferreira M, Oliveira A, Vaz Velho H (2008) Bronchioloalveolar carcinoma associated with congenital pulmonary airway malformation in an asymptomatic adolescent. Revista Portuguesa de Pneumologia 14(2):285–290
Valfre L, Conforti A, Nahom A, Bagolan P (2010) The outcome of expectant management of congenital cystic adenomatoid malformation of the lung. European Journal of Pediatric Surgery 20(6):412 (author reply 412)
van Leeuwen K, Teitelbaum DH, Hirschl RB, Austin E, Adelman SH, Polley TZ, Marshall KW, Coran AG, Nugent C (1999) Prenatal diagnosis of congenital cystic adenomatoid malformation and its postnatal presentation, surgical indications, and natural history. Journal of Pediatric Surgery 34(5):794–798 (discussion 798–799)
Tran H, Fink MA, Crameri J, Cullinane F (2008) Congenital cystic adenomatoid malformation: monitoring the antenatal and short-term neonatal outcome. Aust N Z J Obstet Gynaecol 48(5):462–466
Doros L, Schultz KA, Stewart DR, Bauer AJ, Williams G, Rossi CT, Carr A, Yang J, Dehner LP, Messinger Y, Hill DA (1993) DICER1-related disorders. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews(R). University of Washington, Seattle
Nishibayashi SW, Andrassy RJ, Woolley MM (1981) Congenital cystic adenomatoid malformation: 1 30-year experience. J Pediatr Surg 16(5):704–706
Browdie D, Todd D, Agnew R, Rosen W, Beardmore H (1993) The use of “nonanatomic” pulmonary resection in infants with extensive congenital adenomatoid malformation of the lung. J Thorac Cardiovasc Surg 105(4):732–736
Sapin E, Lejeune V, Barbet JP, Carricaburu E, Lewin F, Baron JM, Barbotin-Larrieu F, Helardot PG (1997) Congenital adenomatoid disease of the lung: prenatal diagnosis and perinatal management. Pediatr Surg Int 12(2–3):126–129
Waszak P, Claris O, Lapillonne A, Picaud JC, Basson E, Chappuis JP, Salle BL (1999) Cystic adenomatoid malformation of the lung: neonatal management of 21 cases. Pediatr Surg Int 15(5–6):326–331
Kim HK, Choi YS, Kim K, Shim YM, Ku GW, Ahn KM, Lee SI, Kim J (2008) Treatment of congenital cystic adenomatoid malformation: should lobectomy always be performed? Ann Thorac Surg 86(1):249–253
Johnson SM, Grace N, Edwards MJ, Woo R, Puapong D (2011) Thoracoscopic segmentectomy for treatment of congenital lung malformations. J Pediatr Surg 46(12):2265–2269
Fascetti-Leon F, Gobbi D, Pavia SV, Aquino A, Ruggeri G, Gregori G, Lima M (2013) Sparing-lung surgery for the treatment of congenital lung malformations. J Pediatr Surg 48(7):1476–1480
Bagrodia N, Cassel S, Liao J, Pitcher G, Shilyansky J (2014) Segmental resection for the treatment of congenital pulmonary malformations. J Pediatr Surg 49(6):905–909. doi:10.1016/j.jpedsurg.2014.01.021
Cagle PT, Thurlbeck WM (1988) Postpneumonectomy compensatory lung growth. Am Rev Respir Dis 138(5):1314–1326. doi:10.1164/ajrccm/138.5.1314
Nakajima C, Kijimoto C, Yokoyama Y, Miyakawa T, Tsuchiya Y, Kuroda T, Nakano M, Saeki M (1998) Longitudinal follow-up of pulmonary function after lobectomy in childhood-factors affecting lung growth. Pediatr Surg Int 13(5–6):341–345
Komori K, Kamagata S, Hirobe S, Toma M, Okumura K, Muto M, Kasai S, Hayashi A, Suenaga M, Miyakawa T (2009) Radionuclide imaging study of long-term pulmonary function after lobectomy in children with congenital cystic lung disease. J Pediatr Surg 44(11):2096–2100
Keijzer R, Chiu PP, Ratjen F, Langer JC (2009) Pulmonary function after early vs late lobectomy during childhood: a preliminary study. J Pediatr Surg 44(5):893–895
Naito Y, Beres A, Lapidus-Krol E, Ratjen F, Langer JC (2012) Does earlier lobectomy result in better long-term pulmonary function in children with congenital lung anomalies? A prospective study. J Pediatr Surg 47(5):852–856
Calvert JK, Lakhoo K (2007) Antenatally suspected congenital cystic adenomatoid malformation of the lung: postnatal investigation and timing of surgery. J Pediatr Surg 42(2):411–414
Pelizzo G, Barbi E, Codrich D, Lembo MA, Zennaro F, Bussani R, Schleef J (2009) Chronic inflammation in congenital cystic adenomatoid malformations. An underestimated risk factor? J Pediatr Surg 44(3):616–619
Conforti A, Aloi I, Trucchi A, Morini F, Nahom A, Inserra A, Bagolan P (2009) Asymptomatic congenital cystic adenomatoid malformation of the lung: is it time to operate? J Thorac Cardiovasc Surg 138(4):826–830
Kim YT, Kim JS, Park JD, Kang CH, Sung SW, Kim JH (2005) Treatment of congenital cystic adenomatoid malformation-does resection in the early postnatal period increase surgical risk? Eur J Cardiothorac Surg 27(4):658–661
Marshall KW, Blane CE, Teitelbaum DH, van Leeuwen K (2000) Congenital cystic adenomatoid malformation: impact of prenatal diagnosis and changing strategies in the treatment of the asymptomatic patient. AJR 175(6):1551–1554
Tsai AY, Liechty KW, Hedrick HL, Bebbington M, Wilson RD, Johnson MP, Howell LJ, Flake AW, Adzick NS (2008) Outcomes after postnatal resection of prenatally diagnosed asymptomatic cystic lung lesions. J Pediatr Surg 43(3):513–517
Rothenberg SS, Kuenzler KA, Middlesworth W, Kay S, Yoder S, Shipman K, Rodriguez R, Stolar CJ (2011) Thoracoscopic lobectomy in infants less than 10 kg with prenatally diagnosed cystic lung disease. J Laparoendosc Adv Surg Tech Part A 21(2):181–184
Albanese CT, Sydorak RM, Tsao K, Lee H (2003) Thoracoscopic lobectomy for prenatally diagnosed lung lesions. J Pediatr Surg 38(4):553–555
Kaneko K, Ono Y, Tainaka T, Sumida W, Kawai Y, Ando H (2010) Thoracoscopic lobectomy for congenital cystic lung diseases in neonates and small infants. Pediatr Surg Int 26(4):361–365
Garrett-Cox R, MacKinlay G, Munro F, Aslam A (2008) Early experience of pediatric thoracoscopic lobectomy in the UK. J Laparoendosc Adv Surg Tech A 18(3):457–459. doi:10.1089/lap.2007.0038
Crombleholme TM, Coleman B, Hedrick H, Liechty K, Howell L, Flake AW, Johnson M, Adzick NS (2002) Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg 37(3):331–338
Yong PJ, Von Dadelszen P, Carpara D, Lim K, Kent N, Tessier F, Delisle MF, Wong T, Blair G, Skarsgard ED (2012) Prediction of pediatric outcome after prenatal diagnosis and expectant antenatal management of congenital cystic adenomatoid malformation. Fetal Diagn Ther 31(2):94–102
Cass DL, Olutoye OO, Cassady CI, Moise KJ, Johnson A, Papanna R, Lazar DA, Ayres NA, Belleza-Bascon B (2011) Prenatal diagnosis and outcome of fetal lung masses. J Pediatr Surg 46(2):292–298
Alamo L, Reinberg O, Vial Y, Gudinchet F, Meuli R (2013) Comparison of foetal US and MRI in the characterisation of congenital lung anomalies. Eur J Radiol 82(12):e860–866
Beydon N, Larroquet M, Coulomb A, Jouannic JM, Ducou le Pointe H, Clement A, Garel C (2013) Comparison between US and MRI in the prenatal assessment of lung malformations. Pediatr Radiol 43(6):685–696
Hubbard AM, Crombleholme TM, Adzick NS, Coleman BG, Howell LJ, Meyer JS, Flake AW (1999) Prenatal MRI evaluation of congenital diaphragmatic hernia. Am J Perinatol 16(8):407–413
Matsuoka S, Takeuchi K, Yamanaka Y, Kaji Y, Sugimura K, Maruo T (2003) Comparison of magnetic resonance imaging and ultrasonography in the prenatal diagnosis of congenital thoracic abnormalities. Fetal Diagn Ther 18(6):447–453
Liu YP, Chen CP, Shih SL, Chen YF, Yang FS, Chen SC (2010) Fetal cystic lung lesions: evaluation with magnetic resonance imaging. Pediatr Pulmonol 45(6):592–600
Winters WD, Effmann EL (2001) Congenital masses of the lung: prenatal and postnatal imaging evaluation. J Thorac Imaging 16(4):196–206
Epelman M, Kreiger PA, Servaes S, Victoria T, Hellinger JC (2010) Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR 31(2):141–157
Loh KC, Jelin E, Hirose S, Feldstein V, Goldstein R, Lee H (2012) Microcystic congenital pulmonary airway malformation with hydrops fetalis: steroids vs open fetal resection. J Pediatr Surg 47(1):36–39
Curran PF, Jelin EB, Rand L, Hirose S, Feldstein VA, Goldstein RB, Lee H (2010) Prenatal steroids for microcystic congenital cystic adenomatoid malformations. J Pediatr Surg 45(1):145–150
Peranteau WH, Wilson RD, Liechty KW, Johnson MP, Bebbington MW, Hedrick HL, Flake AW, Adzick NS (2007) Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn Ther 22(5):365–371
Morris LM, Lim FY, Livingston JC, Polzin WJ, Crombleholme TM (2009) High-risk fetal congenital pulmonary airway malformations have a variable response to steroids. J Pediatr Surg 44(1):60–65
Knox EM, Kilby MD, Martin WL, Khan KS (2006) In-utero pulmonary drainage in the management of primary hydrothorax and congenital cystic lung lesion: a systematic review. Ultrasound Obstet Gynecol 28(5):726–734
Harrison MR, Adzick NS, Jennings RW, Duncan BW, Rosen MA, Filly RA, Goldberg JD, deLorimier AA, Golbus MS (1990) Antenatal intervention for congenital cystic adenomatoid malformation. Lancet 336(8721):965–967
Adzick NS, Harrison MR, Crombleholme TM, Flake AW, Howell LJ (1998) Fetal lung lesions: management and outcome. Am J Obstet Gynecol 179(4):884–889
Hedrick HL, Flake AW, Crombleholme TM, Howell LJ, Johnson MP, Wilson RD, Adzick NS (2005) The ex utero intrapartum therapy procedure for high-risk fetal lung lesions. J Pediatr Surg 40(6):1038–1043 (discussion 1044)
Cass DL, Olutoye OO, Cassady CI, Zamora IJ, Ivey RT, Ayres NA, Olutoye OA, Lee TC (2013) EXIT-to-resection for fetuses with large lung masses and persistent mediastinal compression near birth. J Pediatr Surg 48(1):138–144
Acknowledgement
The authors would like to thank Michel Atlas, Reference and Acquisitions Librarian, Kornhauser Health Sciences Library, University of Louisville, for her assistance with the literature search and citation management. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Downard, C.D., Calkins, C.M., Williams, R.F. et al. Treatment of congenital pulmonary airway malformations: a systematic review from the APSA outcomes and evidence based practice committee. Pediatr Surg Int 33, 939–953 (2017). https://doi.org/10.1007/s00383-017-4098-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-017-4098-z