
Abstract
Aim
To review cases of congenital ureteric stenosis treated in the period between 1999 and 2007. We propose to analyze the type of presentation, management and results.
Material and methods
We report 17 children aged 20 days to 8 years with obstructive uropathy due to congenital stenosis of the ureter at one or more levels. This condition could be mistaken for the more common pelviureteric junction obstruction (PUJO) or primary megaureter, but it is a distinct and more serious anomaly. 13 of the 17 children had one or more associated anomalies, the most significant of which was a contralateral multicystic dysplastic kidney. Other associated anomalies included PUJO, megacalyx, vesicoureteric reflux, urogenital sinus, duplicate vagina, anorectal malformation and agenesis of the bladder. 16 children were symptomatic at presentation, with uremia (serum creatinine >1 mg/dl) in 5, while 1 was diagnosed antenatally. The correct preoperative diagnosis was made in only three children. Reconstruction included ureteroureteral anastomosis, ureteric reimplantation or ureteral substitution.
Results
There is follow up for 15 of the 17 patients. Length of follow up ranges from 1 to 7 years (average 2.7 years). There was satisfactory urinary drainage established in all 17 cases and uremia has resolved 3 of the 5 children. The children with solitary functioning kidney are at risk of uremia in later life.
Conclusion
Congenital ureteric stenosis is a rare condition, but distinct anomaly with possible grave consequence and has been distinguished from other causes of congenital ureteric obstruction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Supravesical causes of congenital obstructive uropathy include pelviureteric junction obstruction (PUJO), primary megaureter, ureteral ectopia and ureterocoele. Apart from these common conditions affecting the uppermost or lowermost portions of the ureter, the main length of the ureter in between its ends is generally not the site of congenital obstruction. Stenosis affecting this segment of ureter is known but rare [1]. We present 17 such children, emphasizing the frequency of the concomitant anomalies, difficulty in diagnosis and the need for innovative reconstructive surgery. The aim of this study is to review cases of congenital ureteric stenosis treated in the period 1999–2007. We propose to analyze the type of presentation, management and results.
Materials and methods
Case material
Seventeen children (10 males, 7 females), age 20 days to 8 years with congenital ureteric stenosis were seen in the period 1999–2007. In all these cases, there was an abrupt change in the ureteric caliber from grossly dilated ureter to very narrow ureter, which would admit a number 3 ureteric catheter with difficulty (Fig. 1). The stenosis affected the ureter at various levels and was multiple in four renal units. While the maximal region of stenosis was usually of short length, the ureter below the stenosis was considered adequately wide and supple for a safe ureteroureteral anastomosis in only three cases. Ureteric stenosis as an isolated anomaly was seen in only 4 of the 17 children, while the remaining 13 had one or more associated anomalies, both in the urinary tract and elsewhere (Fig. 2). Since the presentation and management were often strongly affected by the associated malformations, we have grouped these children according to their pathological anatomy, especially as regards the contralateral renal unit and/or lower urinary tract (Types A–E, Table 1).

Operative photograph demonstrating ureteric stenosis with abrupt change in ureteral caliber. The stenosis just admits a number 3 ureteric catheter

Schematic diagram listing the anomalies found in 17 cases
Management
The diagnosis of ureteric stenosis was established preoperatively by RGP (Fig. 3) in only 3 of the 17 cases. The condition was suspected in two of these three, as there was unilateral hydroureteronephrosis with contralateral MCDK, while in the third, initial imaging clearly demonstrated a hydroureter up to the mid abdomen only (Fig. 4). In the remaining 14 children, the diagnosis of ureteric stenosis was made only during or after an operative intervention. These operative interventions included pyeloplasty (done, failed or abandoned) in seven, ureteric reimplantation (done, or failed) in two, during ureterostomy or undiversion in four and during bladder substitution in one.

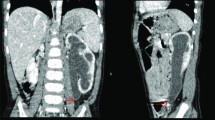
Retrograde pyelogram showing mid ureteric stenosis

Ultrasound and nuclear scan showing dilatation of ureter upto its mid portion
While each individual case had its own diagnostic and management dilemmas, the operative management undertaken in these 17 children is summarized as follows.
Urinary diversion (n = 6)
Urinary diversion in the form of an ureterostomy was employed in six infants, three of whom were uremic (Type B cases) and one had a pyonephrosis. Five have been undiverted while one has been advised nephrectomy for nonfunctioning kidney.
Definitive surgical management
The following surgical techniques were employed. Ureteric reimplantation (n = 9) was possible when the ureteric stenosis was confined to the lower one-third of the ureter and there was sufficient length of dilated ureter above the stenosis to reach the bladder. Psoas hitch of the bladder was employed in two of these cases, while a right reimplant with left to right transureteroureterosotomy was employed in one child with bilateral ureteric stenoses (Type E).
Excision of the stenosed segment and end-to-end ureteral anastomosis (n = 3) was possible when the ureter below the stenosis was considered healthy and supple. In all three cases, a double J stent was placed and removed uneventfully.
Ureteral substitution (n = 4) was employed for long and multiple stenosis. In two children (one Type A, one Type B), the lower ureter was replaced with myotomised appendix [2]. One of these (Type B) had undergone excision of the distal stenosis and reimplantation of the dilated ureter above the stenosis at an earlier operation. He had a solitary functioning left hydronephrotic kidney and on follow up, he was found to have increasing hydronephrosis and renal failure (serum creatinine 4.2 mg/dl). On reexploration multiple stenosis were found in the ureter cranial to the reimplanted segment with dilated ureter in between the stenoses (Fig. 5). This segment of the ureter was replaced with appendix and he was stabilized with a serum creatinine of 1.3 mg/dl. The girl with Type D anomaly underwent replacement of the entire left ureter with the refluxing right ureter (left to right end-to-end pyeloureterostomy with right reimplant) with the right renal pelvis reconnected to the right ureter using an “appendicular bridge”. A fourth ureteral substitution using a Monti tube was used for multiple left ureteric stenoses in a Type E child with bladder agenesis.

Retrograde pyelogram illustrating multiple stenoses (arrows) with ureteric dilatation in between resembling a string of sausages. At initial surgery, only the lower most stenosis was recognized and treated by reimplantation. The stenoses present in the more proximal ureter were recognized only after persistent uremia and hydronephrosis were noted in the follow up. Ureteral replacement with appendix was successfully performed. This cases illustrates the importance of visualizing the entire ureter by retrograde pyelogram even if a single stenosis is seen at surgery
Bladder augmentation/substitution with Mitrofanoff stoma (n = 2). The two girls with Type E required more complicated reconstruction. The child with bilateral ureteric stenoses, female hypospadias, duplex vagina and anorectal malformation was initially managed with right reimplantation and left to right transureteroureterosotomy. She developed retention, urinary infection, increasing hydronephrosis and reflux. After a temporizing vesicostomy, she was reconstructed with ileocystoplasty, Mitrofanoff stoma and redo reimplantation. She remains well (serum creatinine 0.6 mg/dl) on intermittent catherization.
The girl with bladder agenesis underwent bladder substitution with an ileocecal segment with appendicular Mitrofanoff. Her ureter had multiple stenoses in the lower half but the upper dilated right ureter could be reimplanted into the ileocecal bladder substitute. The left ureter had multiple stenoses throughout its length and a Monti tube was used to connect the left renal pelvis to the bladder substitute. She remains stable on intermittent catheterization with a serum creatinine 1 mg/dl.
All the resected specimens were submitted for pathological examination, each revealed stenotic obstruction (Fig. 1) and no case of ureteral valve was seen.
Results
Follow-up data is available for 15 of 17 children. Length of follow-up ranges from 1 to 7 years (average 2.7 years). All children have been relieved of their presenting symptoms. None has palpable masses or urinary infection. Two girls are on intermittent catheterization for lower urinary tract problems. Of the total of five children in renal failure preoperatively (all Type B), the mean serum creatinine has decreased from a preoperative value of 3.44 mg/dl (range 1.3–5.4 mg/dl) to a follow-up value 1.16 mg/dl (range 0.5–2.2 mg/dl) after reconstruction, though three children continue to have raised though stable creatinine ranging from 1.1 to 2.2 mg/dl. The Type D child has shown deterioration of renal function (0.9 vs. 2.4 mg/dl). Improvement in differential function in Type A was variable and follow-up values were available in five of six cases. In three children, differential renal function in the affected kidney improved or stabilized (0, 46, 59% preoperative vs. 35, 45.4, 61% postoperative) while in two, the renal function deteriorated (36, 18% preoperative vs. 18, 4% postoperative) either due to pyonephrosis or due to renal dysplasia (multiple cysts). This deterioration could be also be due to the contralateral healthy kidney undergoing compensatory hypertrophy.
Discussion
Congenital ureteric stenosis affecting one or more regions of the ureter is a rare cause of hydroureteronephrosis and has to be distinguished from other more common supravesical ureteric obstructions such as primary megaureter, ectopic ureter and ureterocoele. In our opinion, congenital ureteric stenosis is possibly a forme fruste ureteral atresia and is associated with a high degree of obstruction to the affected renal unit, unlike primary megaureter, which often has a benign course. Evidences for this conclusion are diminished split renal function (mean 28%) in cases with contralateral normal kidney (Type A), raised serum creatinine in most Type B cases, palpable hydronephrosis, urinary ascites, ipsilateral renal dysplasia (multiple renal cysts) and the frequent association with contralateral MCDK (Fig. 2). Of additional interest is the association with bladder agenesis and a variant of cloaca in two cases and also a hand anomaly in another case (Fig. 2).
A definitive preoperative diagnosis was generally not made in the majority of our cases because the condition was not suspected. An abrupt cessation of ureteric dilatation generally cranial to the juxtavescial region is demonstrable in ultrasound, intravenous pyelogram, radio nucleotide scan and by retrograde pyelography. Magnetic resonance urography, though not done in our cases, would have been useful to demonstrate radiologically the abrupt cessation of the ureteral dilation along the course of the ureter. The presence of contralateral MCDK is a strong pointer to the diagnosis. In an article reviewing urological anomalies with solitary kidneys [3], true renal agenesis was associated with more anomalies than MCDK. However, in the series reported herein, we find that contralateral ureteral stenosis is an important and potentially lethal association with MCDK.
Treatment is surgical and there is little place for expectant management as all but one child were symptomatic and some were in renal failure. Surgical techniques were needed to be individualized and include ureteroureteral anastomosis, ureteric reimplantation and ureteral substitution after removing the obstructing segment. Uremic infants need to be stabilized with ureterostomy. Recovery from renal failure was good in childhood but it remains to be seen whether this will be maintained in adolescence. Recovery of differential renal function with a normal contralateral kidney was variable.
Most congenital urological anomalies are currently diagnosed antenatally [4, 5] and expectant management is advised for most PUJO and primary megaureters with a benign outcome. Congenital ureteric stenosis, however, is not a benign entity and can mimic PUJO and primary non-refluxing megaureter. In conclusion, we have described how to suspect, diagnose and treat this condition based on our experience with 17 children.
References
Hwang AH, McAleer IM, Shapiro E, Miller OF, Krous HF, Kaplan GW (2005) Congenital mid ureteric strictures. J Urol 174:1999–2002. doi:10.1097/01.ju.0000176462.56473.0c
Dagash H, Sen S, Chacko J, Karl S, Ghosh D, Parag P, Mackinnon AE (2008) The appendix as ureteral substitute: a report of 10 cases. J Pediatr Urol 4(1):14–19. doi:10.1016/j.jpurol.2007.08.004
Kaneyama K, Yamataka A, Satake S, Yanai T, Lane GJ, Kaneko K, Yamashiro Y, Miyano T (2004) Associated urological anomalies in children with solitary kidney. J Pediatr Surg 39(1):85–87. doi:10.1016/j.jpedsurg.2003.09.010
Cauchi JA, Chandran H (2005) Congenital ureteric strictures: an uncommon cause of antenatally detected hydronephrosis. Pediatr Surg Int 21:556–568. doi:10.1007/s00383-005-1455-0
Gitlin J, Kaefer M (2002) Congenital mid ureteral stricture presenting as prenatal hydronephrosis. J Urol 168:1154–1156. doi:10.1016/S0022-5347(05)64615-0
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kannaiyan, L., Karl, S., Mathai, J. et al. Congenital ureteric stenosis: a study of 17 children. Pediatr Surg Int 25, 513–517 (2009). https://doi.org/10.1007/s00383-009-2368-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-009-2368-0