
Abstract
Purpose
A defect in a phosphate-regulating gene leads to the most common form of rickets: X-linked hypophosphatemic rickets (XLH) or vitamin D-resistant rickets (VDDR). XLH has been associated with craniosynostosis, the sagittal suture being the most commonly involved.
Methods
We present three patients with rickets and symptomatic sagittal suture craniosynostosis all of whom presented late (>2 years of age). Two had a severe phenotype and papilledema, while the third presented with an osseous bulging near the anterior fontanel and experienced chronic headaches.
Results
All underwent successful cranial vault expansion.
Conclusions
Rachitic patients with scaphocephaly should be screened for craniosynostosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Craniosynostosis can be associated with a metabolic disorder and/or deficiency. Rickets and iatrogenic etiologies (i.e., thyroid replacement therapy for hypothyroidism) are most likely [1–3]. X-linked hypophosphatemic rickets (XLH), also known as vitamin D-resistant rickets (VDDR), is the most common metabolic cause of craniosynostosis. This disorder affects 1:20,000 persons and is caused by a loss of function mutation in a phosphate-regulating gene (PHEX) [4, 5]. Craniosynostosis secondary to rickets is rare and often has a late presentation accompanied by signs of elevated intracranial pressure (ICP) such as headaches, vomiting, papilledema, or bulging of the anterior fontanel [6–8]. In a study of 59 children under age 9 with rickets due to any cause, one third had craniosynostosis and three required surgery [9]. The presentation and management of craniosynostosis in patients with rickets differs somewhat from common non-metabolic causes and issues specific to rachitic forms should be addressed. Symptomatic craniosynostosis is more likely to occur when treatment for the rickets is absent or inadequate; consequently, radiologic screening is recommended for newly discovered XLH patients not diagnosed in early infancy [7, 8, 10].
We present three patients with craniosynostosis and rickets. In each case, there was a delay in diagnosis and lack of recognition of the clinical signs or symptoms indicating elevated intracranial pressure. The objectives of this report are to draw attention to the need for careful cranial screening of all patients with rickets, provide some additional perspective on the metabolic basis for this association, and discuss management of rachitic craniosynostosis.
Case reports
Patient 1
A 3 year-old female was referred to our clinic with papilledema, severe scaphocephaly, and CT-proven of sagittal synostosis. She was the product of a full-term uncomplicated pregnancy and in good health. She walked independently at 15 months but bowing of the legs led to subsequent evaluation and confirmation of XLH rickets. There was no family history of rickets and her condition had been managed with calcitriol and sodium potassium phosphate supplementation.
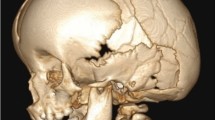
She had a long and thin head shape that caregivers presumed was a normal consequence of her rickets. At age 3 years, an ophthalmologist was concerned about elevation of the optic nerve heads on exam and ordered a CT scan that revealed fusion of the sagittal suture and physiologic closure of the metopic suture. There was also significant endocortical scalloping. (Fig. 1) These findings prompted referral to our center. Physical examination showed a short but otherwise healthy-appearing little girl with severe scaphocephaly (cranial index = 0.65; normal 0.75). Her head circumference was 50.5 cm (75th percentile). She was asymptomatic, but an ophthalmologic exam revealed farsightedness with anisometropia and bilateral papilledema. Operative intervention to expand the cranium was recommended.

(Patient 1) 3D-reformatted axial CT showing fusion of the sagittal suture and classic scaphocephalic head shape (a, b); axial image with severe endocortical erosion (scalloping) (c)
She underwent prompt operative decompression and total calvarial remodeling and expansion to improve her cranial shape. Removal of the bone confirmed complete absence of subdural cerebrospinal fluid, thinning of the dura, and significant endocortical erosion and focal calvarial thinning. The procedure increased her overall head circumference 3 cm and she had a post-operative cephalic index of 0.74. She recovered uneventfully and subsequent ophthalmologic examination 6 months after the procedure showed complete resolution of the papilledema and improvement in the anisometropia.
Patient 2
This boy was born to a mother with XLH and there was a strong maternal family history of this disorder. He was diagnosed with XLH at age 6 months and was managed with calcitriol and sodium potassium phosphate supplementation. His head shape was scaphocephalic from birth, but this finding did not prompt diagnostic testing for craniosynostosis until 2 years of age. CT scan at that time demonstrated fusion of the sagittal suture and physiologic metopic closure. There was no appreciable subdural space and significant endocortical erosion and thinning.
At presentation, the child was noted to have significant frontal bossing, severe scaphocephaly (cranial index 0.62), and a large head circumference (52.5 cm, 98th percentile). The patient had no symptoms commonly associated with elevated intracranial pressure, but there was bilateral papilledema on ophthalmologic exam. Based on these findings, the patient underwent total calvarial remodeling and expansion. During procedure, the bone was found softer than usual, and there was complete obliteration of the subdural space with endocortical scalloping. The operative expansion increased the head circumference 3.5 cm, and the post-operative cephalic index was 0.72. Recovery was without incident, and he required no further intervention. The preoperative papilledema resolved on subsequent ophthalmologic assessment.
Patient 3
This girl was diagnosed with XLH at age 1 year and was managed with standard therapy. Her head shape was unremarkable in infancy, but she developed a prominent bony elevation in the area of the anterior fontanelle that was noticed at age 3 years. She also had complained of occasional headaches. She was referred to our center for evaluation.
On presentation, she had an almost normal cranial index of 0.73 and a head circumference 50 cm (60th percentile). Besides her diminished height and bony prominence in the region of the anterior fontanelle, her head shape and physical exam were unremarkable. CT scan was ordered and demonstrated fusion of the sagittal suture, decreased pericerebral space, and erosion of the inner surface of the cranium consistent with elevated ICP (Fig. 2). There was no evidence of papilledema on ophthalmologic exam. Nevertheless, given her symptoms and CT findings the family elected for cranial expansion. The combination of CT findings, i.e., decreased pericerebral space, endocortical erosion, and the frontal prominence in the area of the anterior fontanelle (“volcano” sign) are highly suggestive of pressure. Whether the pressure was developing slowly from birth due to a congenital synostosis or whether this developed in a delayed fashion related to the medical treatment remains unknown.

(Patient 3) 3D-reformatted axial CT showing fusion of the sagittal suture and classic scaphocephalic head shape (a, b); axial image with severe endocortical erosion (scalloping) (c)
Intraoperative findings include significant thinning of the bone and dura, marked endocortical scalloping, and absence of the subdural cerebrospinal fluid that resolved after removal of the bone. She recovered from the operation uneventfully and her headaches resolved. Her head circumference increased 2 cm and the cephalic index improved to 0.78. Post-operative ophthalmologic exam showed no papilledema.
Discussion
Norwegian pediatrician Olga Immerslund first described the association between XLH rickets and craniosynostosis in 1951 [11]. However, Heschl is credited for reporting the first patients with nutritional (vitamin D deficiency) rickets and craniosynostosis in 1873 [12]. While most physicians assume that nutritional rickets has been eradicated, this disease is still widely present in impoverished areas of the world. It is less prevalent in developed countries, but the prevalence of vitamin D deficient foods and greater indoor habits have led to an unexpected re-emergence [1]. Wang and colleagues presented a recent example of craniosynostosis secondary to dietary rickets [13]. Some medications that limit the absorption of vitamin D can also lead to this disorder. Shetty and coworkers reported a case of a 10-month-old male with hypophosphatemia secondary to chronic therapy with a phosphate-binding antacid containing magnesium and aluminum hydroxides. The child developed typical skeletal features of rickets including craniotabes, frontal bossing, rachitic rosary, epiphyseal widening, and craniosynostosis of the sagittal, lambdoid, and squamosal sutures [14].
The pathogenesis of craniosynostosis in rickets has been studied in the X-linked hypophosphatemic mouse model (HYP) and illustrates the differences between PHEX mutations versus fibroblast growth factor receptor mutations seen in other forms of craniosynostosis [15, 16]. Two key pathways of the bone mineralization are implicated in the pathogenesis of craniosynostosis in HYP. PHEX dysfunction in HYP mice leads to increased circulating fibroblast growth factor 23 (FGF23), a protein that increases elimination of phosphate in the urine [17, 18]. FGF23 crossbinds to FGF receptor 2 and 3 according to Murthy [19]. PHEX has also been shown to interact with a matrix extracellular phosphoglycoprotein (MEPE) leading to downstream inhibition of bone mineralization. Therefore, defective PHEX may lead to disturbances in bone mineralization via interactions with FGF 23 and MEPE. This leads to deregulation of phosphate balance and the inhibition of bone mineralization respectively, making the child highly susceptible to craniosynostosis [20].
XLH rickets result from a loss of function mutation in the phosphate regulating gene with homologies to endopeptidases (PHEX) on the human X chromosome (Xp22.2-p22.1) that reduces the reabsorption of phosphate in the renal tubule. Increased excretion of phosphate impairs bone mineralization and leads to osseous softening or osteomalacia. Patients can present with dental caries, short stature, and various orthopedic problems such as lower limb deformities (i.e., genu varum or valgus). Various opthalmologic findings include proptosis, optic nerve head elevation, and papilledema—the latter two problems may be secondary to intracranial pressure elevations caused by craniosynostosis. XLH patients require close attention and multiple treatments from a composite care team consisting of endocrinology, orthopedic, ophthalmology, craniofacial surgery, neurosurgery, nephrology, and dental care services. XLH rickets is treatable with carefully monitored simultaneous calcitriol and phosphate intake; however, adequately treated patients may still develop secondary problems such as nephrocalcinosis [2–7, 9, 13, 21].
Identifying the presence of craniosynostosis in the XLH sub-population can be challenging. In one series of 24 rickets patients, radiological and clinical data was reviewed and 13 were found to have sagittal synostosis [22]. While 10 patients had the classic scaphocephalic head shape (and thus could be suspected based on phenotype), three patients had normal shaped heads. The cranial vault configuration in the scaphocephalic patients was noted to be slightly different from the non-syndromic forms and both clinical and radiological monitoring was recommended in this group. Other reviews have suggested that boys with XLH may be more at risk for craniosynostosis than girls [13]. Although the sagittal suture is more commonly involved, the involvement of the coronal sutures in addition to the expected sagittal involvement has been described. The symptomatic craniosynostotic phenotype in XLH and other types of rickets varies somewhat (Table 1). This is likely due to variability in the severity of disease and/or individual differences in response to treatment, compliance, and detection of disease [24]. Along with multiple suture involvement comes an increase in ICP as documented by Tamburrini et al. [25]. Without multiple suture involvement, the ICP increase may not be clinically obvious as seen in case 3 of this series. The authors would argue that despite a lack of measured ICP elevation, a measured value may not be prognostic outside of the age at which the measurement is computed, and that surgical decision-making based upon highly suggestive CT findings is justified. That is, a normal pressure at 3 years of age does not portend normal pressure in the future after further growth. Cranial expansion allows for the treatment of elevated ICP if present, and the increased intracranial volume should help to prevent ICP issues in the near future.
Operative management of patients with XLH can present some challenges. The severity of the cranial deformity (e.g., significant frontal bossing) requires a more extensive reconstruction than would be needed in typical forms of scaphocephaly. Because these patients present late, the cranial bone is usually thicker and more difficult to contour [26]. Moreover, the presence of severe endocortical scalloping, seen in all of our patients, can increase the risk of dural tearing during initial bone removal. Finally, altered calcium concentrations can be present and have been reported in XLH patients undergoing cranial vault reconstruction. These issues should be taken into account when choosing the type of repair and during reconstructive craniectomy [24, 27].
Conclusions
All patients with XLH rickets should be repeatedly assessed for the presence or development of craniosynostosis. Routine ophthalmologic examination in these patients has been advocated to identify papilledema, but this test is not sensitive in young children and is a late finding [6, 21]. Because the sagittal suture is most commonly involved and often results in a predictable head shape, we recommend several simple anthropometric parameters to screen for this condition: head circumference percentile over 90 %, a cephalic index of less than 0.75, and a bony elevation over the anterior fontanel. Any child with these findings should undergo CT evaluation. According to and consistent with Virchow’s Law, a worsening phenotype is expected with any synostosis as long as there is cranial growth. The authors argue that although two-dimensional and planar measures can be helpful in identifying craniosynostosis, they do not capture the totality of cranial shape changes in sagittal synostosis. Some patients with sagittal synostosis have a normal cranial index due to compensatory bi-temporal bulging, but are very narrow and pinched in the vertex above the squamosal sutures. For these reasons, qualitative CT features are a necessary aspect of phenotypic evaluation.
References
Lowdon J (2011) Rickets: concerns over the worldwide increase. J Fam Health Care 21(2):25–9
Panchal J, Uttchin V (2003) Management of craniosynostosis. Plast Reconstr Surg 111(6):2032–48
Penfold JL, Simpson DA (1975) Premature craniosynostosis—a complication of thyroid replacement therapy. J Pediatr 86(3):360–3
Carpenter TO (1997) New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin North Am 44(2):443–466
Pitt MJ (1991) Rickets and osteomalacia are still around. Radiol Clin North Am 29(1):97
Glass LR, Dagi TF, Dagi LR (2011) Papilledema in the setting of x-linked hypophosphatemic rickets with craniosynostosis. Case Rep Ophthalmol 2:376–381
Glorieux FH (1991) Rickets, the continuing challenge. N Engl J Med 325:1875–1877
Willis FR, Beattie TJ (1997) Craniosynostosis in X-linked hypophosphaetemic rickets. J Paediatr Child Health 33:78–79
Reilly BJ, Leeming JM, Fraser D (1964) Craniosynostosis in the rachitic spectrum. J Pediatr 64:396–405
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M (1991) Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med 325(26):1843–8, 26
Immerslund O (1951) Craniostenosis and vitamin D-resistant rickets. Acta Paediatr 40:449–451
Heschl M (1873) Einige Bemerkunger über Föntale und prä-mature obliterationen der Schädelnähte. Vjscher Prakt Heilk 120:135
Wang PI, Marcus JR, Fuchs HE, Mukundan S Jr (2007) Craniosynostosis secondary to rickets: manifestations on computed tomography. Radiol Case Rep 2:43 [Online]
Shetty AK, Thomas T, Rao J (1998) Rickets and secondary craniosynostosis associated with long-term antacid use in an infant. Arch Pediatr Adolesc Med 152:1243–1245
Baumgartner JE, Seymour-Dempsey K, Teichgraeber JF, Xia JJ, Waller AL, Gateno J (2004) Nonsynostotic scaphocephaly: the so-called sticky sagittal suture. J Neurosurg 101(1 Suppl):16–20
Roy WA, Iorio RJ, Meyer GA (1981) Craniosynostosis in vitamin D-resistant rickets. A mouse model. J Neurosurg 55(2):265–71
Ranch D, Zhang MY, Portale AA, Perwad F (2011) Fibroblast growth factor 23 regulates renal 1,25-dihydroxyvitamin D and phosphate metabolism via the MAP kinase signaling pathway in Hyp mice. J Bone Miner Res 26(8):1883–90
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H et al (2003) Human fibroblast growth factor-23 mutants suppress Na + -dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem 278(4):2206–11
Murthy AS (2009) X-linked hypophosphatemic rickets and craniosynostosis. J Craniofac Surg 20(2):439–42
Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM, Mundy GR et al (2005) Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: a model for impaired mineralization in X-linked rickets (HYP). Bone 36(1):33–46
Tuite GF, Chong WK, Evanson J, Narita A, Taylor D, Harkness WF et al (1996) The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurg 38(2):272–8
Currarino G (2007) Sagittal synostosis in X-linked hypophosphatemic rickets and related diseases. Pediatr Radiol 37(8):805–12
Inman, Page C. B.S.; Mukundan, Srinivasan Jr Ph.D., M.D.; Fuchs, Herbert E. Ph.D., M.D.; Marcus, Jeffrey R. M.D. Craniosynostosis and Rickets. Plast Reconstr Surg. 2008 Apr;121(4):217e-8e
Freudlsperger C, Hoffmann J, Castrillion-Oberndorfer G, Engel M (2013) Bilateral coronal and sagittal synostosis in X-linked hypophosphatemic rickets: A case report. J Craniomaxillofac Surg [Published online ahead of print 7 March 2013] http://www.sciencedirect.com/science/article/pii/S1010518213000553 Accessed April 15 2013
Tamburrini G, Caldarelli M, Massimi L, Santini P, Di Rocco C (2005) Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst 21:913–921
Seruya M, Oh AK, Boyajian MJ, Myseros JS, Yaun AL, Keating RF, Rogers GF (2013) Age at initial consultation for craniosynostosis: comparison across different patient characteristics. J Craniofac Surg Jan 24(1):96–8
Garg R, Khanna P, Pandia M (2010) Anaesthetic considerations in a child with rickets and craniosynostosis for linear strip craniectomy and frontal advancement. Indian J Anaesth 54(4):350–1
Acknowledgments
We thank Dr. Martin Bazan for his assistance with the preparation of the figures and the table
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None
Rights and permissions
About this article

Cite this article
Jaszczuk, P., Rogers, G.F., Guzman, R. et al. X-linked hypophosphatemic rickets and sagittal craniosynostosis: three patients requiring operative cranial expansion: case series and literature review. Childs Nerv Syst 32, 887–891 (2016). https://doi.org/10.1007/s00381-015-2934-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2934-9