
Abstract
Purpose
Neurocutaneous melanosis (NCM) is a rare congenital disorder occurring in children born with multiple or large congenital melanocytic nevi (CMN) in association with melanocytic deposits in the leptomeninges. Multiple associations between NCM and other syndromes or neurologic abnormalities have been reported. Of note, there exists a possible association between NCM and tethered cord (TC).
Methods
We retrospectively reviewed charts and films of all patients with the diagnosis of NCM at the Children’s Hospital of Pittsburgh (CHP) from August 2002 to present.
Results
Five children met the criteria for NCM at our institution over a 12-year period. Apart from the melanocytic deposits, one or more additional spinal abnormalities were identified in all children. Three children had radiographic evidence of a low-lying conus medullaris, two of which also demonstrated lipomatous infiltration of the filum terminale, consistent with a tethered cord (TC).
Conclusions
Clinical features of NCM include dermatologic and neurologic manifestations. To date, this is the first series to note an association between NCM and TC. While nearly all recent series of NCM patients advocate early MRI of the neuroaxis, we recommend screening imaging of the spine on children with possible NCM regardless of the locations of CMN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurocutaneous melanosis (NCM) is a rare congenital disorder occurring in children born with multiple or large congenital melanocytic nevi (CMN) in association with melanocytic deposits in the leptomeninges. It was initially described by Rokitansky in 1861, who reported a teenager with large cutaneous nevi and hydrocephalus with leptomeningeal deposits discovered at autopsy [33]. In NCM, the abnormal proliferation of melanocytes can be seen in the leptomeninges, brain, or spinal cord parenchyma. NCM is thought to represent an error of migration of melanoblasts [22, 19].
NCM was formally defined in 1972 by Fox and colleagues as multiple or giant pigmented nevi in patients without malignant transformation of the cutaneous lesions and without malignant deposits elsewhere than the leptomeninges [14]. While the leptomeningeal lesions are most often benign, they have been reported to undergo malignant transformation in nearly 3 % of patients [15]. The following diagnostic criteria were proposed by Kadonaga and Frieden in 1991 and remain in clinical use today: (1) large or multiple congenital melanocytic nevi in association with meningeal melanosis or melanoma, (2) no evidence of cutaneous melanoma, except in patients in whom the examined areas of meningeal deposits are found to be benign, and (3) no evidence of meningeal melanoma, except in patients in whom the examined areas of cutaneous nevi are benign [22].
Multiple associations between NCM and other syndromes or neurologic abnormalities have been reported. NCM has been seen in conjunction with other neurocutaneous disorders, including neurofibromatosis and Sturge-Weber syndrome [10]. Approximately 8–10 % of children with NCM have cysts in the posterior fossa, including arachnoid cysts and Dandy-Walker malformation [3, 23, 25, 32]. Hydrocephalus is common [32]. Other intracranial findings include lissencephaly, Chiari I malformation, middle cranial fossa arachnoid cyst, and encephalocraniocutaneous lipomatosis [10, 21, 6, 2]. A recent study reported that a significant proportion of these patients have epilepsy and developmental delay [32]. Associated spinal abnormalities include arachnoid cysts, spinal lipomas, and syringomyelia [24, 34]. To date, there is only one report of a tethered spinal cord in a patient with NCM [13]. We describe our single-institution experience with pediatric NCM and note the association with tethered cord (TC).
Methods
The charts and films of all patients with the diagnosis of NCM at the Children’s Hospital of Pittsburgh (CHP) from August 2002 to present were reviewed. The diagnosis of NCM was made in accordance with the aforementioned criteria of Kadonaga and Frieden, as determined by the pediatric neurooncologist at CHP (author RJ). Contrast-enhanced MR imaging (MRI) of the brain and spine was obtained on all patients and reviewed by a pediatric neuroradiologist. Neurologic assessment was performed by a pediatric neurosurgeon and/or pediatric neurologist. Developmental delay was defined as failure to meet appropriate milestones. Low-lying conus medullaris was defined as the position of the tip of the conus medullaris below the L2–L3 interspace. Medical charts were reviewed to assess clinical and radiographic characteristics including age at diagnosis of NCM, gender, radiographic location of melanocytic deposits, other cranial and/or spinal abnormalities (posterior fossa cysts, hydrocephalus, spinal cord tethering, others), need for and type of surgical intervention, duration of follow-up, and clinical status at most recent evaluation. Institutional review board approval was obtained prior to data collection.
Results
Five children met the criteria for NCM at our institution over a 12-year period. All children were male. At present, four children are alive; one child died from progression of his disease. Age at diagnosis ranged from birth to 5 years. All children displayed multifocal melanocytic deposits on neuroimaging; the child who died had diffuse leptomeningeal involvement throughout his brain and spinal cord, while one other child also has diffuse involvement. Follow-up ranged from 18 months to 14 years. Clinical features of the five children are summarized in Table 1.
Regions of intracranial involvement included the thalamus, cerebellar hemispheres, pons, and deep mesial structures, including the amygdala. A supratentorial arachnoid cyst was present in one child, and one child had a retrocerebellar cyst. Two children had evidence of hydrocephalus requiring placement of a ventriculoperitoneal shunt.
Apart from the melanocytic deposits, one or more additional spinal abnormalities were identified in all children. Two children had evidence of cervicothoracic arachnoid cysts and diffuse spinal leptomeningeal involvement. One child had scoliosis that required surgical correction. Three children had a low-lying conus medullaris, and two of these were accompanied by lipomatous infiltration of the filum terminale.
At the time of initial diagnosis, three children were symptomatic. One child was born with hydrocephalus and suffered from the progressive loss of neurologic function and pain throughout his life secondary to the diffuse and aggressive nature of his disease. A second child presented at age 2.5 years with symptoms of myelopathy related to extensive spinal involvement and syringomyelia. A third child presented with developmental delay and scoliosis. Two children remain asymptomatic at present.
Of the cohort, four children required surgical intervention for a neurologic problem. One child required treatment of hydrocephalus with fenestration of a posterior fossa cyst and placement of a ventriculoperitoneal shunt. Another child underwent surgical biopsy of a diffuse intradural, extramedullary spinal abnormality causing syringomyelia. One child underwent orthopedic instrumentation for correction of scoliosis. Another child underwent prophylactic release of an asymptomatic TC. Of the four children who are alive, two are neurologically and developmentally normal, one child is delayed and also carries the diagnosis of autism, and one child has a seizure disorder controlled on medication.
Case series
Case one
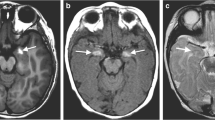
A full-term male infant was noted to have multiple CMNs, including diffuse, large lesions located below the nipple line on the dorsum of the trunk (Fig. 1). He had an uncomplicated prenatal course. An MRI of the brain and total spine performed on day of life (DOL) 7 demonstrated T1 hyperintense/T2 hypointense abnormalities of the amygdala, bilateral optic tracts, optic chiasm, anterior pons, inferior medulla, and bilateral cerebellar hemispheres. Thin rims of melanin at the cortical surface of the bilateral frontal and parietal lobes were noted. The child had a mega cisterna magna and enlarged ventricles (Fig. 2). Spinal MRI showed a thoracic arachnoid cyst (Fig. 3), and the conus medullaris positioned at the inferior aspect of the L2 vertebral body.

Multiple CMNs, including diffuse, large lesions located below the nipple line on the dorsum of the trunk

Coronal T1-weighted MRI (left) showing signal abnormality in the cerebellar hemispheres and sagittal T2-weighted MRI (right) demonstrating posterior fossa cyst

Cervicothoracic T2-weighted MRI showing an arachnoid cyst dorsal to the spinal cord
This child underwent open surgical fenestration of his posterior fossa cyst on DOL 36, and placement of a ventriculoperitoneal shunt (VPS) for persistent ventriculomegaly on DOL 41. He suffered rapid clinical progression of his disease. An MRI obtained at 3 months of age revealed a thickened, fat-infiltrated filum terminale, with the conus medullaris positioned at the L2–L3 interspace. At 8 months of age, the conus medullaris was positioned at the L3–L4 interspace with increased thickening of the filum terminale and a significant increase in leptomeningeal enhancement. Given the rapid and debilitating nature of his NCM, the family decided not to pursue TC release.
At 15 months of age, he presented with 5 days of paraplegia and refractory pain. Neuroaxis imaging revealed complete effacement of the intracranial subarachnoid space. There was radiographic progression of his spinal disease with diffuse deposits filling the thecal sac, with evidence of hemorrhage and spinal cord compression. At 17 months, the family elected to offer the child palliative care, and he died shortly thereafter. Autopsy showed multiple metastases. A detailed description of the genetic, histological, and other biological features is underway (manuscript in preparation).
Case two
A full-term male infant was noted at birth to have multiple CMNs after an uncomplicated prenatal course. On DOL 1, the child underwent an MRI of the brain and total spine which showed a Dandy-Walker variant, hydrocephalus, and a pontine T1 hyperintense/T2 hypointense lesion. No enhancement was noted. The conus medullaris was positioned at the inferior portion of the L2 vertebral body; no other spinal abnormalities were noted.
On DOL 2, the patient underwent placement of a frontal VPS. He manifested seizure activity post-operatively as evident by limb shaking. This was confirmed with EEG, and anti-epileptic medication (phenobarbital) was initiated. The seizures were easily controlled on oral agents. At 2.5 years of age, he developed right leg weakness and a foot drop. A repeat MRI of the neuroaxis revealed no change in the brain lesion, but the interval development of an arachnoid cyst ventral to the cord from C4 to T2, a syrinx in the low thoracic spinal cord extending to the conus medullaris, clumping of the cauda equina, and enhancement of the right L4 nerve root. The conus medullaris now terminated at the level of the L3 vertebral body (Fig. 4). He was taken to the operating room and underwent osteoplastic laminotomies for biopsy of the enhancing lesions and fenestration of the syrinx into the subarachnoid space. The pathology revealed well-differentiated melanocytes (staining positive for S-100 and HMB-45), with a Ki-67 index of <3 %.

T2-weighted sagittal (left) and axial (right) thoracolumbar MRI showing low-lying conus medullaris
At 10 years of age, the patient developed severe lower back pain with a radicular component into both lower extremities. MRI showed an increase in the size of the holocord septated syrinx. He was started on gabapentin for neuropathic pain, as well as sorafenib. Within a few weeks, his pain improved significantly and he tolerated weaning of gabapentin. Six months later, MRI showed a significant decrease in the size of the syrinx. The patient is 11 years old at present and has a mild right foot drop, which is improving with physical therapy.
Case three
A full-term male infant was documented at birth to have CMN covering more than 50 % of the body surface. He was large for gestational age, with an otherwise uncomplicated prenatal course. On DOL 4, MRI of the brain showed enhancement of the right hippocampus without other abnormalities, and spine imaging was found to be normal.
The child was subsequently diagnosed with developmental delay and autism by a pediatric neurologist. He was found to have severe scoliosis and required surgical correction at age 10 years. Spinal imaging showed a conus medullaris positioned at L1, without fatty infiltration of the filum terminale, syrinx, or arachnoid cyst. His scoliosis was hypothesized to be secondary to extensive dermal scarring from multiple cutaneous excisions of nevi and skin grafting.
The child received serial imaging over the next 5 years of life, without evidence of radiographic progression. At age 15, brain imaging showed a new area of enhancement in the left thalamic region; this has remained stable over the last 1.5 years, and the child has no clinical sequelae.
Case four
A male twin was noted at birth to have multiple CMN. His initial MRI was performed at 5 months of age and revealed enhancing lesions in the right amygdala and left cerebellar hemisphere. The conus medullaris terminated at the level of L3, with lipomatous infiltration of the filum terminale (Fig. 5).

T2-weighted sagittal (left) MRI showing low-lying conus medullaris and T1-weighted axial (right) MRI showing fatty infiltration of filum terminale
The child underwent neurosurgical evaluation for the radiographic TC, and the parents requested prophylactic release of the TC. This was performed without complication at 18 months of age. At the time of surgery, the child had no neurological sequelae secondary to tethering and could ambulate well but was not yet toilet-trained.
Further imaging to date has shown no progression of the intracranial lesions. The patient is 25 months old at present and without neurologic deficit.
Case five
A male infant was born at home and noted by his parents to have CMN at birth. The parents brought the child to medical attention because a close relative had been diagnosed with CMN. The family requested an MRI for the evaluation of NCM. An MRI of the brain and spine performed at 5 months of age revealed enhancing lesions of the right amygdala and right cerebellar hemisphere, a retrocerebellar cyst without hydrocephalus, and enhancement of the leptomeninges surrounding the conus medullaris. The conus medullaris was positioned at the L1-2 interspace.
The patient is 16 months old at present and remains asymptomatic. Interval imaging at 10 and 16 months of age has revealed no radiographic progression of disease.
Discussion
NCM is a rare, congenital, non-hereditary syndrome, believed to be the result of faulty embryonic neuroectodermal migration. Giant (defined as >20 cm in adults and, in infants and neonates, >9 cm diameter on the head and 6 cm on the body) or multiple CMNs are required for the diagnosis of NCM. Age at initial presentation can vary, and while most cases are associated with cutaneous melanocytic deposits at birth, the disease can manifest throughout childhood and, rarely, in adulthood. The clinical course of NCM is also highly variable, as is evident in our cohort, ranging from those who are asymptomatic long-term survivors to those with a rapid course and death in infancy or early childhood [7].
Clinical features of NCM include dermatologic and neurologic manifestations. Two thirds of patients with NCM have large congenital nevi and one third have multiple, but no giant, nevi (defined as three or more lesions) [22, 29]. Children with large or multiple congenital nevi at highest risk for NCM have nevi located in the head and neck regions or over the thoracic spine [22, 29]. Melanocytic deposition in the central nervous system most commonly affects the mesial temporal structures, amygdala, cerebellum, and pons, and up to 20 % of patients have spinal involvement, including leptomeningeal thickening, arachnoiditis, and syringomyelia [13, 10, 31, 4].
The risk of developing symptomatic manifestations of NCM has not been fully elucidated. Previously, patients with NCM brought to medical attention were biased towards those children with neurologic symptoms. Retrospective reviews have suggested the incidence of symptomatic NCM, based on large cohorts of patients with CMN, to range from 2.5–11.4 % [5, 9]. Symptoms of NCM usually manifest before the age of 2 and include headache, focal seizures, developmental delay, intracranial hemorrhage, cranial neuropathy, and myelopathy or radiculopathy [31, 3]. It is reported that nearly two thirds of children develop hydrocephalus and symptoms secondary to elevated intracranial pressure [21, 29].
TC describes the clinical condition resulting from pathologic tissue attachments that limit movement of the spinal cord, often associated with an abnormally short and/or thickened filum terminale [18, 1, 17]. TC can occur as a result of congenital or acquired conditions, including myelomeningocele (MM), lipomyelomeningocele, split cord malformation, dermal sinus tract, tight filum terminale, spinal lipoma, neurentic cyst, and others. Non-congenital causes include postsurgical scar, most commonly after MM repair; infectious or traumatic injuries can also result in tethering [12, 1, 17]. TC is often diagnosed by the presence of a low-lying conus medullaris, defined as a conus medullaris terminating below the L2 vertebral body. Importantly, the conus medullaris relatively ascends within the spinal canal to its final adult position at L2 by 3–4 months of age [17], and not all TCs are associated with a low-lying conus medullaris [35, 36, 16].
To date, there is only one mention of TC in a child with NCM in the literature [13]. In a series of 11 children with NCM who underwent MRI of the spine, Foster and colleagues report one child (an incidence of 9 %) with radiographic findings of TC, but do not mention subsequent management. The authors advocated that all infants with a giant nevus involving the lumbosacral region undergo a spinal MRI. While our series is small, there was a high association between NCM and TC, with three of five children showing evidence of tethering. From an embryological standpoint, this association is plausible. It is believed that NCM is a failure of proper migration of neural crest cells [22, 32], and TC is a failure of secondary neurulation [17]. Broadly, both entities likely represent a failure of normal neurocutaneous morphogenesis.
Of note, the child in our series who underwent release of his TC was asymptomatic and neurologically normal. The role of surgery in asymptomatic patients with radiographic tethering remains unclear [8, 26, 30, 28, 27]. At our institution, release of radiographic tethering in asymptomatic children is offered if the children have multiple radiographic findings (low-lying conus medullaris, filum terminale lipoma, syrinx).
Before the introduction of MRI, it was known that spinal leptomeningeal involvement in NCM could lead to spinal cord damage and myelopathy, as evident at surgery and autopsy [11]. Nearly all recent series of patients with NCM advocate early MRI of the neuroaxis, and the non-invasive diagnosis of NCM cannot be made without such imaging. Humphreys recommended MRI in patients with large or medium-sized lumbosacral nevi [20]. A recent study by Ramaswamy and colleagues recommended baseline imaging within the first year of life for children with possible NCM [32]. This recommendation was based on the high incidence of spinal arachnoid cysts and cerebral parenchymal deposits. The authors advocated imaging in the first 4 months of life because of the possibility that myelination may obscure melanin deposits. Again, given the high association with TC in our series and the fact that the conus medullaris does not assume its final position until about 3 months of age, we support imaging of the complete neuroaxis at 3–4 months of age in the absence of symptoms of hydrocephalus or focal neurologic deficit. The one other report of TC in association with NCM in the literature [13] described a child with a large cutaneous nevus in the region of the lumbar spine. While cutaneous stigmata can be a hallmark of an underlying neural tube abnormality such as TC, large lumbosacral nevi were not present in each child with TC in our series; therefore, we recommend screening imaging on children with possible NCM regardless of the locations of CMN.
Abbreviations
- NCM:
-
Neurocutaneous melanosis
- CMN:
-
Congenital melanocytic nevus
- MRI:
-
Magnetic resonance imaging
- TC:
-
Tethered cord
- DOL:
-
Day of life
- VPS:
-
Ventriculoperitoneal shunt
- EEG:
-
Electroencephalography
References
Agarwalla PK, Dunn IF, Scott RM, Smith ER (2007) Tethered cord syndrome. Neurosurg Clin N Am 18(3):531–547. doi:10.1016/j.nec.2007.04.001
Ahmed I, Tope WD, Young TL, Miller DM et al (2002) Neurocutaneous melanosis in association with encephalocraniocutaneous lipomatosis. J Am Acad Dermatol 47(2 Suppl):S196–S200
Alikhan A, Ibrahimi OA, Eisen DB (2012) Congenital melanocytic nevi: where are we now? Part I. Clinical presentation, epidemiology, pathogenesis, histology, malignant transformation, and neurocutaneous melanosis. J Am Acad Dermatol 67(4):495.e1–17. doi:10.1016/j.jaad.2012.06.023, quiz 512–494
Barkovich AJ, Frieden IJ, Williams ML (1994) MR of neurocutaneous melanosis. AJNR Am J Neuroradiol 15(5):859–867
Bittencourt FV, Marghoob AA, Kopf AW, Koenig KL et al (2000) Large congenital melanocytic nevi and the risk for development of malignant melanoma and neurocutaneous melanocytosis. Pediatrics 106(4):736–741
Chaloupka JC, Wolf RJ, Varma PK (1996) Neurocutaneous melanosis with the Dandy-Walker malformation: a possible rare pathoetiologic association. Neuroradiology 38(5):486–489
Chu WC, Lee V, Chan YL, Shing MM et al (2003) Neurocutaneous melanomatosis with a rapidly deteriorating course. AJNR Am J Neuroradiol 24(2):287–290
Cornette L, Verpoorten C, Lagae L, Van Calenbergh F et al (1998) Tethered cord syndrome in occult spinal dysraphism: timing and outcome of surgical release. Neurology 50(6):1761–1765
DeDavid M, Orlow SJ, Provost N, Marghoob AA et al (1997) A study of large congenital melanocytic nevi and associated malignant melanomas: review of cases in the New York University Registry and the world literature. J Am Acad Dermatol 36(3 Pt 1):409–416
Demirci A, Kawamura Y, Sze G, Duncan C (1995) MR of Parenchymal neurocutaneous melanosis. AJNR Am J Neuroradiol 16(3):603–606
Faillace WJ, Okawara SH, McDonald JV (1984) Neurocutaneous melanosis with extensive intracerebral and spinal cord involvement. Report of two cases. J Neurosurg 61(4):782–785. doi:10.3171/jns.1984.61.4.0782
Filippidis AS, Kalani MY, Theodore N, Rekate HL (2010) Spinal cord traction, vascular compromise, hypoxia, and metabolic derangements in the pathophysiology of tethered cord syndrome. Neurosurg Focus 29(1):E9. doi:10.3171/2010.3.FOCUS1085
Foster RD, Williams ML, Barkovich AJ, Hoffman WY et al (2001) Giant congenital melanocytic nevi: the significance of neurocutaneous melanosis in neurologically asymptomatic children. Plast Reconstr Surg 107(4):933–941
Fox H, Emery JL, Goodbody RA, Yates PO (1964) Neuro-cutaneous melanosis. Arch Dis Child 39:508–516
Hale EK, Stein J, Ben-Porat L, Panageas KS et al (2005) Association of melanoma and neurocutaneous melanocytosis with large congenital melanocytic naevi—results from the Nyu-Lcmn Registry. Br J Dermatol 152(3):512–517. doi:10.1111/j.1365-2133.2005.06316.x
Hendrick EB, Hoffman HJ, Humphreys RP (1983) The tethered spinal cord. Clin Neurosurg 30:457–463
Hertzler DA 2nd, DePowell JJ, Stevenson CB, Mangano FT (2010) Tethered cord syndrome: a review of the literature from embryology to adult presentation. Neurosurg Focus 29(1):E1. doi:10.3171/2010.3.FOCUS1079
Hoffman HJ, Hendrick EB, Humphreys RP (1976) The tethered spinal cord: its protean manifestations, diagnosis and surgical correction. Childs Brain 2(3):145–155
Humes RA, Roskamp J, Eisenbrey AB (1984) Melanosis and hydrocephalus. Report of four cases. J Neurosurg 61(2):365–368. doi:10.3171/jns.1984.61.2.0365
Humphreys RP (1996) Clinical evaluation of cutaneous lesions of the back: spinal signatures that do not go away. Clin Neurosurg 43:175–187
Kadonaga JN, Barkovich AJ, Edwards MS, Frieden IJ (1992) Neurocutaneous melanosis in association with the Dandy-Walker complex. Pediatr Dermatol 9(1):37–43
Kadonaga JN, Frieden IJ (1991) Neurocutaneous melanosis: definition and review of the literature. J Am Acad Dermatol 24(5 Pt 1):747–755
Kang SG, Yoo DS, Cho KS, Kim DS et al (2006) Coexisting intracranial meningeal melanocytoma, dermoid tumor, and Dandy-Walker cyst in a patient with neurocutaneous melanosis. Case report. J Neurosurg 104(3):444–447. doi:10.3171/jns.2006.104.3.444
Kasantikul V, Shuangshoti S, Pattanaruenglai A, Kaoroptham S (1989) Intraspinal melanotic arachnoid cyst and lipoma in neurocutaneous melanosis. Surg Neurol 31(2):138–141
Kim KH, Chung SB, Kong DS, Seol HJ et al (2012) Neurocutaneous melanosis associated with Dandy-Walker complex and an intracranial cavernous angioma. Childs Nerv Syst 28(2):309–314. doi:10.1007/s00381-011-1638-z
Koyanagi I, Iwasaki Y, Hida K, Abe H et al (1997) Surgical treatment supposed natural history of the tethered cord with occult spinal dysraphism. Childs Nerv Syst 13(5):268–274
Liptak GS (1995) Tethered spinal cord: update of an analysis of published articles. Eur J Pediatr Surg 5(Suppl 1):21–23. doi:10.1055/s-2008-1066257
McLone DG, La Marca F (1997) The tethered spinal cord: diagnosis, significance, and management. Semin Pediatr Neurol 4(3):192–208
Mena-Cedillos CA, Valencia-Herrera AM, Arroyo-Pineda AI, Salgado-Jimenez MA et al (2002) Neurocutaneous melanosis in association with the Dandy-Walker complex, complicated by melanoma: report of a case and literature review. Pediatr Dermatol 19(3):237–242
Oakes WJ (1996) The borderlands of the primary tethered cord syndrome. Clin Neurosurg 43:188–202
Pavlidou E, Hagel C, Papavasilliou A, Giouroukos S et al (2008) Neurocutaneous melanosis: report of three cases and up-to-date review. J Child Neurol 23(12):1382–1391. doi:10.1177/0883073808319069
Ramaswamy V, Delaney H, Haque S, Marghoob A et al (2012) Spectrum of central nervous system abnormalities in neurocutaneous melanocytosis. Dev Med Child Neurol 54(6):563–568. doi:10.1111/j.1469-8749.2012.04275.x
Rokitansky J (1861) An excellent case of a pigmented mole with effused pigmentation of the inner meninges of the brain and spinal cord [in German]. Allg Wein Mediz 26:113–116
van Heuzen EP, Kaiser MC, de Slegte RG (1989) Neurocutaneous melanosis associated with intraspinal lipoma. Neuroradiology 31(4):349–351
Warder DE, Oakes WJ (1993) Tethered cord syndrome and the conus in a normal position. Neurosurgery 33(3):374–378
Warder DE, Oakes WJ (1994) Tethered cord syndrome: the low-lying and normally positioned conus. Neurosurgery 34(4):597–600, discussion 600
Acknowledgments
We would like to thank Ashok Panigrahy, M.D., for contributing images for this manuscript.
Conflict of interest
The authors have no financial disclosures.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tian, A.G., Foster, K.A., Jakacki, R.I. et al. Neurocutaneous melanosis is associated with tethered spinal cord. Childs Nerv Syst 31, 115–121 (2015). https://doi.org/10.1007/s00381-014-2526-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-014-2526-0