
Abstract
Introduction
Currently, there are only a few reported cases of symptomatic or asymptomatic subpial (intramedullary) spinal lipoma, and therefore no guidelines are available to indicate surgery. These lesions are infrequently associated with spina bifida.
Case report
Herein, we provide our experience in the neurosurgical intervention of compressive myeloradiculopathy for encephalocraniocutaneous lipomatosis (ECCL). The patient initially presented with bilateral upper hand paralysis, then regained muscle power after surgery and during 1 year of follow-up. We discuss the neurosurgical indications and intervention, imaging studies, other associated symptoms, and the pathogenesis of ECCL in an infant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Encephalocraniocutaneous lipomatosis (ECCL), also named as Fishman's syndrome and Haberland syndrome, is a rare, congenital, neurocutaneous disorder [3, 16]. It manifests clinically by unilateral lipomatous hamartomas of the scalp, eyelid, and outer globe of the eye. Naevus psiloliparus is the most common skin anomaly and is considered the hallmark of ECCL [6, 10]. Porencephalic cysts, cortical atrophy, cranial asymmetry, marked developmental delay, mental retardation, congenital heart malformations, lytic bone lesions, hypospadias, and cryptorchidism may also exist [6, 10, 16]. Since the first case reported in 1970 by Haberland and Perou, roughly 60 cases have been described, and diagnostic criteria have been developed and revised [13]. Several cases have been reported with regards to cutaneous and visceral involvement [6], as well as the association with diseases such as fibrodysplasia [4], neurofibromatosis [8, 12], papillary glioneuronal tumors [15], and low-grade astrocytomas [5]. Neurosurgical intervention was discussed in a case report on placing a ventricular shunt and debulking of central nervous system lipomas [2]. However, few articles have reported surgical interventions for symptomatic intraspinal subpial lipomas. To the best of our knowledge, this is the first article to report surgical decompression of ECCL presenting with compressive cervical myeloradiculopathy leading to a significant improvement in upper limb motor function in an infant.
Case report
This girl was born to a G3P2 mother at 41 + 2 weeks of gestation via normal spontaneous delivery with a body weight at birth of 2,900 g. There were no abnormalities during pregnancy, no complications during delivery, and the child's prenatal and neonatal history was unremarkable. Only maternal condyloma accuminatum was noted during delivery.
After birth, multiple progressive enlarging soft tissue masses were noted over her right face, deltoid muscle, and inguinal area. Fat tissue was favored by ultrasound examination. The activity and muscle power were grossly normal initially after birth. However, bilateral upper limb weakness starting from the right side gradually developed after 1 week (Medical Research Council grade 0). A physical examination revealed absent grasp reflex and Moro reflex. A laboratory examination showed no abnormalities except for mild liver dysfunction. Screening for TORCH showed negative results. An ophthalmologic examination showed left eye absent light reflex and iris coloboma with suspected choroidal coloboma over the temporal upper periphery. The right pupil presented with normal light reflex, however, there was dermolipoma of the right limbus and mild abducens palsy. A neonatal auditory test showed an absent response of the right ear with brain stem response: right 55 dB, left 25 dB.
Due to bilateral upper limb paralysis, nerve conduction velocity/electromyography (EMG) was performed. Nerve conduction studies of bilateral upper limbs were normal. Needle EMG revealed marked active denervation with reduced recruitment of motor unit action potentials in bilateral deltoid, biceps, extensor digitorum communis, abductor pollicis brevis, and cervical paraspinal muscles. Electrophysiological findings were suggestive of a widespread intraspinal canal lesion involving the C5 through T1 level.
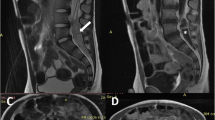
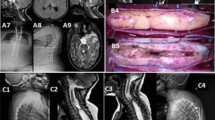
Brain and cervical spine magnetic resonance imaging (MRI) was therefore performed, and showed a large lipomatous lesion extending from C0/1 to T9/10, with compression to the spinal cord and brain stem; however, there was no evidence of hydrocephalus. A small homogenous lipomatous lesion was also seen in the right cerebellopontine angle (Fig. 1). Due to the complete motor dysfunction of bilateral upper limbs, we performed suboccipital craniectomy and C1 to T9 laminotomy for compressive cervical myeloradiculopathy. Intraoperatively, the lipoma was found to be located intramedullary at the dorsal site, from C0 to T9/10 level (Fig. 2). The intramedullary lipoma was partially removed from its central part for decompression and to preserve the nerve roots under intraoperative neuromonitoring. The histopathological report was a lipoma composed of an abnormal collection of mature adipose tissue, and no malignancy was noted.

Lipomatosis in the right petrous bone and right cerebellopontine angle

Intraoperative findings: the lipoma was found to be located intramedullary at the dorsal site, from C0 to T9/10 level
After the operation, she gradually regained the muscle power in bilateral hands over a 1-week period, from grade 0 preoperatively to grade 3. During an outpatient follow-up visit 1 month later, she was able to move her hands freely. Follow-up EMG 4 months later revealed less prominent fibrillation density in all denervated muscles. Nine months after the operation, she was able to move both of her hands freely with full muscle strength and full range of motion. She met the normal developmental milestones and there was no scoliosis.
Discussion
According to the revised diagnostic criteria proposed by Moog et al. in 2009 [13], the diagnosis of ECCL is based on the involvement of different systems including the eyes, skin, central nervous system, and others. In the current case, the dermolipoma of the right eye, nevus psiloliparus (hairless scalp lesions) on her scalp, and intraspinal lipoma met the major criteria. Minor criteria such as patchy or streaky non-scarring alopecia (without fatty nevus), subcutaneous lipomas in the frontotemporal region and genital area were also present (Fig. 3). The lesions were located predominantly on the right side, and only iris coloboma was noted in her left eye. The clinical presentation met the criteria of definite ECCL, in that: “three systems were involved, with major criteria in more than two”. The diagnosis of definite ECCL was therefore confirmed. Spinal cord lipomas are a major diagnostic criterion of ECCL, and both intra- and extramedullary lipomas have been reported. Most of the time, they are asymptomatic, and surgery is usually not required [1, 2, 12]. For those who undergo an operation, no neurological deficits have been attributable to spinal cord compression prior to surgery. Two cases reportedly underwent decompressive laminectomy of the cervical spine successfully without neurological deficits [1, 12]. One case with the initial presentation of low back pain without neurological deficits underwent resection of T10-11 lipoma; however, he was paraplegic after the operation [4, 7]. Another case presented with scoliosis and sleep apnea, and received posterior fossa craniectomy, C1-6 laminectomies, and debulking of a cervicomedullary junction lipoma. There was no muscle weakness before or after the operation [2].

The typical cutaneous findings that met the diagnostic criteria of ECCL. a Choristoma and coloboma of the right eye. b Nevus psiloliparus on her scalp. c Cutaneous lipomas on her inguinal area
In the current case, the initial presentation was bilateral upper limb total paralysis, and absence of Moro and grasp reflexes. A lipomatous spinal lesion from C0 to T9 was found in MRI (Fig. 4a and b). Due to compressive cervical myeloradiculopathy, suboccipital craniectomy and C1 to T9 laminotomy were performed. Despite the preoperative MRI showing that most lesions seemed to be extramedullary with clear demarcation, we found intraoperatively that the lipoma was located subpially. The lipoma was closely entangled with the spinal cord. Using intraoperative neuromonitoring, we performed only partial debulking of the lipoma in areas where it was possible to distinguish between the spinal cord and lipomatous tissue under microscopy. The function and muscle power of bilateral hands dramatically improved two days after the operation, from total paralysis to freely movable.

Preoperative MR imaging studies of the brain and cervical spine demonstrated extensive lipoma compression of the cervical cord. a Sagittal T2-weighted image showing a lipoma extending from the medulla oblongata to lower thoracic area. b Axial T1-weighted image of the cervical spine at the level indicated in panel A with a lobulated lipoma that seemed to be intradural and extramedullary to the right side of the spinal cord. Postoperative MR imaging demonstrated enlargement of the lipoma. c Sagittal T2-weighted image, and d Axial T1-weighted image of the cervical spine at the C5-6 level indicated in panel c showed enlargement of the lipoma with mild extraspinal protrusion. The patient remained symptom free
To avoid damage of the spinal cord, we do not suggest complete resection of subpial lipomas. Similar to other reported cases [2], even though the preoperative findings may suggest that the lipomas are extramedullary, they are mostly located intramedullary. Debulking surgery and decompressive laminectomy can greatly assist symptomatic cervical myeloradiculopathy; however, they should be performed with extreme caution. There are only a few reported cases of symptomatic or asymptomatic spinal lipoma, and therefore no guidelines are available to indicate surgery. From the experience of this case, early surgical decompression with careful debulking may be helpful in cases with neurological deterioration.
The MRI in the third and ninth months after the operation showed a residual lipomatous lesion extending from the cervicomedullary junction to about the T9 level, and progression of the lesion size was noted (Fig. 4c and d). However, despite the progression of the lesion seen on MRI, the muscle power of bilateral hands remained good and showed continuing improvement. This may imply that the enlargement of the lipomatous lesion paralleled the growth of the patient, or that the spinal canal was more expansible after the laminotomy allowing for growth of the lesion without causing symptoms.
The neurological manifestation of ECCL includes developmental delay, seizure and mental retardation. Among all of the reported cases, epileptic seizures were reported in almost half of the patients [9, 13, 14]. In the current case, there were no seizures and no developmental delay at the last follow-up. However, long-term follow-up is suggested to detect further abnormal neurodevelopment [10]. Hydrocephalus has been reported in one third of patients with ECCL, and several cases have reported successful placement of ventriculoperitoneal shunts [12, 14]. A tethered cord, lipomyelomeningocele and scoliosis have also been reported as indications for surgery [2]. In the current case, no hydrocephalus, tethered cord, or lipomyelomeningocele were observed. Longitudinal observation for the possible development of scoliosis is needed, however, CNS anomalies such as asymmetric atrophy of hemispheres and calcification have been found in patients with ECCL. This has been proposed to be related to vascular anomalies, which were found in approximately 10 of 54 cases in one review [13]. Progressively dilating vessels leading to subarachnoid hemorrhage was also reported in one case [3]. Currently, there are no recommendations for routine surveillance imaging studies for possible vascular anomalies, and in the current case, there were no vascular anomalies present (by MRI angiography). However, appropriate vascular imaging studies may be needed if new neurological deficits develop.
The pathogenesis of ECCL remains unknown. It is thought to be a sporadic disease resulting from somatic mosaicism [6, 10]. Dysgenesia of the cephalic neural crest and the anterior neural tube is the most widely accepted theory of the pathogenesis of ECCL. No evidence currently exists of genetic transmission or chromosomal abnormalities. Mutations of several genes such as the NF1 allele, genes involving the RAS-MAPK pathway, or vasculogenesis may be involved [11, 13]. In the current case, the cutaneous manifestation followed the Blaschko line in distribution, supporting the idea of mosaicism as the pathogenesis of ECCL.
Choristoma and coloboma of the right eye, nevus psiloliparus on her scalp, cutaneous lipomas on her right temporal face and inguinal area, and intraspinal and intracranial lipomas were typical and common findings that met the diagnostic criteria for ECCL [6]. Other abnormalities reported in previous reports such as brain anomalies, scoliosis, odontoma, vascular aneurysm, epilepsy, or developmental delay [2, 5, 6, 8–10, 12] were not found in the current case.
Conclusion
This is the first article to report that partial debulking of an intraspinal lipoma in an infant with ECCL presenting with compressive cervical myeloradiculopathy led to significant improvements in upper limb motor function, and this improvement persisted despite continuous growth of the spinal lipoma on imaging studies. Hence, we suggest early spinal decompression in symptomatic ECCL cases with myelopathy. With meticulous intra-operative monitoring, patients can benefit greatly, even with partial debulking
References
Alfonso ILP, Cullen RF Jr, Martin-Jimenez R, Bejar RL (1986) Spinal cord involvement in encephalocraniocutaneous lipomatosis. Pediatr Neurol 2(6):380–384
Ayer REZA (2011) Encephalocraniocutaneous lipomatosis: a review of its clinical pathology and neurosurgical indications. J Neurosurg Pediatr Sep 8(3):316–320
Brumback RALR (1987) Fishman's syndrome (encephalocraniocutaneous lipomatosis): a field defect of ectomesoderm. J Child Neurol 2(3):168–169
Deda GÇH, Yavuzer G, Arasıl T (2001) Encephalocraniocutaneous lipomatosis associated with iris coloboma, chorioretinitis and spinal cord involvement: a case report. Brain Dev 23:355–358
Delfino LNFG, Quattrocchi CC, Aiello C, Menchini L, Devito R, Zama M, Claps D, Vigevano F, Longo D (2011) Encephalocraniocutaneous lipomatosis (ECCL): neuroradiological findings in three patients and a new association with fibrous dysplasia. Am J Med Genet A 155A(7):1690–1696
Dhouib A, Hanquinet S, La Scala GC (2013) encephalocraniocutaneous lipomatosis: magnetic resonance imaging findings in a child. J Pediatr S0022–3476(13):00115–00117
Fitoz SAC, Erden I, Akyar S (2002) Intracranial lipoma with extracranial extension through foramen ovale in a patient with encephalocraniocutaneous lipomatosis syndrome. Neuroradiology 44:175–178
Gawel J, Schwartz RA, Jozwiak S (2003) Encephalocraniocutaneous lipomatosis. J Cutan Med Surg 7:61–65
Hauber K, Warmuth-Metz M, Rose C, Br cker E-B, Hamm H (2003) Encephalocraniocutaneous lipomatosis: a case with unilateral odontomas and review of the literature. Eur J Pediatr 162:589–593
Kim DH, Park SB, Lee Y, Im M, Seo YJ, Choi SH, Lee JH (2012) Encephalocraniocutaneous lipomatosis without neurologic anomalies. Ann Dermatol 24(4):476–478
Legius EWR, Eyssen M, Marynen P, Fryns JP, Cassiman JJ (1995) Encephalocraniocutaneous lipomatosis with a mutation in the NF1 gene. J Med Genet 32(4):316–319
Moog UJM, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB (2007) Brain anomalies in encephalocraniocutaneous lipomatosis. Am J Med Genet A 143A(24):2963–2972
Moog U (2009) Encephalocraniocutaneous lipomatosis. J Med Genet 46(11):721–729
Parazzini CTF, Russo G, Mastrangelo M, Scotti G (1999) Encephalocraniocutaneous lipomatosis: complete neuroradiologic evaluation and follow-up of two cases. AJNR Am J Neuroradiol 20(1):173–176
Phi JHPS, Chae JH, Wang KC, Cho BK, Kim SK (2010) Papillary glioneuronal tumor present in a patient with encephalocraniocutaneous lipomatosis: case report. Neurosurg 67(4):E1165–E1169, Am J Med Genet A Jul;155A(7):1690–6
Rubegni PRM, Sbano P, Buonocore G, Perrone S, Fimiani M (2003) Encephalocraniocutaneous lipomatosis (Haberland syndrome) with bilateral cutaneous and visceral involvement. Clin Exp Dermatol 28(4):387–390
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chiang, CC., Lin, SC., Wu, HM. et al. Clinical manifestation and neurosurgical intervention of encephalocraniocutaneous lipomatosis—a case report and review of the literature. Childs Nerv Syst 30, 13–17 (2014). https://doi.org/10.1007/s00381-013-2252-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-013-2252-z