
Abstract
Purpose
Controlled cortical impact (CCI) is commonly used in adult animals to study focal traumatic brain injury (TBI). Our study aims to further study injury mechanisms in children and variable models of pathology in the developing brain.
Methods
Develop a focal injury model of experimental TBI in the immature, postnatal days (PND) 7 and 17 rats that underwent a CCI at varying depths of deflection, 1.5–2.5 mm compared with sham and then tested using the Morris water maze (MWM) beginning on post-injury day (PID) 11. Histopathologic analysis was performed at PID 1 and 28.
Results
In PND 7, the 1.75- and 2.0-mm deflections (diameter (d) = 3 mm; velocity = 4 m/s; and duration = 500 ms) resulted in significant MWM deficits while the 1.5-mm injury did not produce MWM deficits vs. sham controls. In PND 17, all injury levels resulted in significant MWM deficits vs. sham controls with a graded response; the 1.5-mm deflection (d = 6 mm; velocity = 4 m/s; and duration = 500 ms) produced significantly less deficits as compared WITH the 2.0- and 2.5-mm injuries. Histologically, a graded injury response was also seen in both ages at injury with cortical and more severe injuries, hippocampal damage. Cortical contusion volume increased in most injury severities from PID 1 to 28 in both ages at injury while hippocampal volumes subsequently decreased.
Conclusions
CCI in PND 7 and 17 rat results in significant MWM deficits and cortical histopathology providing two different and unique experimental models of TBI in immature rats that may be useful in further investigations into the mechanisms and treatments of pediatric TBI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Head trauma remains a significant pediatric health problem, with an estimated incidence of 230/100,000 [39–41] leading to 100,000–200,000 new cases of pediatric traumatic brain injury (TBI) each year US Census Bureau estimate; (www.census.gov/population/estimates). While it has been estimated that nearly 20–30,000 children are permanently disabled from TBI annually, few experimental models exist. To date, the approach to the care of children with TBI has been derived from the adult guidelines [14]. In the “Guidelines for the Acute Medical Management following Severe Traumatic Brain Injury in Infants, Children, and Adolescents,” by Adelson et al. [2], there were few studies that were significant enough to dictate standards of care. While the clinical management differences in the acute setting between adults and children are obvious, and while there are extensive differences between the immature, developing brain and the mature brain, anatomically, functionally, physiologically, molecularly, etc., there is little understanding of the potential unique response of the immature brain to traumatic injury, both experimentally and clinically [1, 36, 37, 47].
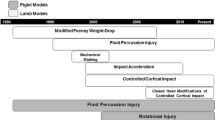
The literature characterizing and describing TBI has for the most part been concentrated in the adult rat [18, 22, 49], including controlled cortical impact (CCI) [18] which has become useful for investigations into the pathophysiology and treatment of TBI [15–17, 19, 29, 31, 58]. Previous studies in children have highlighted that while children as a whole have better outcomes than adults [45], younger children (<4 years), and particularly those <2 years of age, have worse outcomes than older school-age children [42, 43]. While this may be a function of types of injury rather than mechanism per se, [34] and may represent a continuum of injury and response, it has never been tested using rigorous clinical and preclinical methods as experimental models have been lacking. Based on these clinical observations and consistent with our previous work [3, 4], our primary goal has been to develop models of TBI in the immature rat severe enough to produce a functional deficit that could later be studied as to the mechanisms that might contribute to that behavioral deficit and might then translate to the clinical, human condition. Similarly, since the immature brain is changing throughout the developmental cycle, different age at injury models are necessary to adequately study the contributory secondary mechanisms that may be age related and potentially unique after TBI, leading to therapeutic interventions and preclinical studies prior to pediatric clinical trials.
There have been a number of descriptions of TBI models in immature animals to date [5, 6, 8, 20, 21, 27, 30–32, 46, 57, 58], that have begun to define the differing response of the immature brain including diffuse impact acceleration focal injury by weight-drop [27, 32], fluid percussion injury in rats [56], and focal injury in pigs [8, 20] and mice [21]. Although diffuse injury is more common than focal TBI in children [13], parenchymal injury or contusion following TBI plays a significant role in the morbidity and mortality in children [26].
Since a number of different ages at injury have been used across a variety of CNS insults in immature rats including both TBI and ischemia, two spectra of ages that have been commonly used have been postnatal days (PND) 7 and 17. PND 7 rats have been used in a myriad of studies to model the neonatal occurrence of the GABA switch and marked vulnerability of the PND 7 to excitotxocitic injury from NMDA receptor activation in the early work [48]. Indeed the vast majority of studies using the Rice Vanucci model of neonatal hypoxia ischemia have been carried out in PND 7 rats [59]. PND 17 rats have been generally used to model a toddler since this represents the age with maximal number of synapses and cerebral blood flow [52, 53]. There are established models of ischemia utilizing this age at injury [23].
Because of common use of the adult model of CCI [18], in this study we sought to modify our adult CCI model as initially described by Dixon et al. [18] and preliminarily reported on in immature (PND 17) rats [10, 11, 25, 33, 35], and by others [21, 54, 60] to better understand the mechanistic response of the immature brain to traumatic injury and at different ages at injury that resulting in functional deficits as well as quantifiable histologic injury [6]. The present study describes two specific ages at injury models of experimental TBI defining the resultant Morris water maze (MWM) dysfunction following the injury and then secondarily, defining the histologic injury across graded injury levels in PND 7 and 17 rats.
Materials and methods
Subjects
A total of 124 PND 7 and 17 male Sprague Dawley (Harlan, Indianapolis, IN) rat pups (weight—PND 7, 15–20 g and PND 17, 33–40 g) were used to assess the functional and histologic effects of injury following CCI. The subjects were housed with their dams until PND 21 when they were weaned and housed in separate cages. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (protocol # 0009975).
Surgical procedures
The rats were initially anesthetized with 1.5–2.0 % isoflurane in N2O/O2 (2:1) via nosecone. Core temperature was maintained at 37–37.5 °C throughout the procedure using a heating pad and monitored by a rectal temperature probe. The scalp was shaved and prepared with betadine. All the animals for both PND 7 and 17 rats had their heads secured using ear pins in a stereotactic head frame to avoid movement. Using sterile technique, the skull was exposed by a midline sagittal incision and the scalp reflected. Using a high-speed air drill, a 4.0- (PND 7) or 7.0-mm (PND 17) diameter craniotomy was made in the left parietal bone (0.2 mm from midline, 0.2 mm posterior to coronal suture, and 0.2 mm anterior to lambdoid suture, extending laterally to the superior temporal line) under an operating microscope. The bone flap was removed leaving the dura intact.
Controlled cortical impact injury
We have previously described CCI procedures in adult rats [18, 29] and reported its application to the immature rat [7, 11, 25, 33, 35, 64]. A focal cortical impact was delivered at a controlled velocity of 4 m/s, duration of 500 ms, using a 3- or 6-mm tip (PND 7 or 17, respectively). Once the device was positioned, anesthesia was discontinued until the toe pinch reflex returned and then the preset injury delivered. The injury deflection depths for the PND 7 rats were 1.5, 1.75, and 2.0 mm and for the PND 17, 1.5, 2.0, and 2.5 mm vs. sham (craniotomy, no impact). Preliminary studies using deflections of 2.5 and 3.0 mm in the PND 7 and 17, respectively, and the 6-mm tip in the PND 7, lead to excessive tissue damage, and high mortality and therefore were not pursued (PD Adelson, personal communication, 2010). Immediately following injury, anesthesia was resumed, the bone flap replaced, secured with Koldmount dental cement (Vernon-Benshoff, Albany, NY), and the wound sutured. The rats were then transferred to a warmed recovery cage and observed for at least 30 min. After 30 min, and ensuring the resumption of normal gait, the rat pups were transferred back to their litters and observed intermittently over the next 30 min to ensure acceptance by their dams.
Morris water maze testing
The MWM paradigm is an open-field procedure in which rats learn to find a submerged platform to escape from a forced swimming task [51]. We have previously utilized this paradigm to assess functional deficits after diffuse injury in both PND 7 and 17 rats [3, 4]. A large circular tank (180 cm in diameter and 45 cm high) containing water (temperature maintained at 26 ± 1 °C) to a height of 30 cm contains a transparent circular platform (10 cm in diameter and 29 cm high) located in a fixed position in the tank 45 cm from the tank wall and 1 cm below the water surface. Extra-maze visual cues aid the rat in locating the escape platform. Water temperature was maintained at 26 ± 1 °C.
Three separate spatial memory assessments were employed to evaluate learning-acquisition deficits in the immature rats. First, the animals underwent cognitive performance evaluation by placing them in the MWM de novo on post-injury day (PID) 11 without prior training or exposure to the MWM. The subjects were given 4 trials/day for five consecutive days with a hidden platform (n = 10 for each age at injury and each injury severity level). The rats started a trial once from each of four randomized starting locations within the tank, placed in the water against and facing the tank wall, and then released to swim freely about the tank in order to find the hidden platform, up to 120 s. If the rat was unable to locate the platform within the allotted time, it would be manually directed to the platform and placed on it if necessary. The rat would be allowed to remain on the platform for 30 s, then removed and placed in a dry, warmed holding incubator between trials. After a 4-min intertrial interval, the animal was placed back in the maze for the next trial. On the fifth day of MWM testing (PID 15), the rats underwent a probe trial where the platform was removed from the maze and the time spent in the “target quadrant” (the quadrant where the platform was previously located) was measured with a Chromatrak video-tracking system (San Diego Instruments, San Diego, CA) for 120 s. This was then compared with the time spent in the remaining three quadrants (collectively referred to as “out of quadrant”). Lastly, on PID 16–17, the platform was raised to 1 in. above the water level making it visible to the rat (visual probe trial). Each animal was released from each of the four randomized starting positions to locate, swim to, and mount the visible platform to evaluate nonspecific visual deficits as a potential factor affecting the rats’ ability to locate the platform. Latency to finding the submerged platform was the primary dependent variable for cognitive performance. Throughout each testing period, all subjects were observed for signs of distress or inability to swim via remote camera.
Histologic analysis
Histologic analysis of the injury was evaluated at PID 1 and 28 (n = 120 with n = 10 at each age at injury and each injury severity level). Rats were anesthetized with isoflurane in oxygen and transcardially perfusion fixed with 4 % buffered formalin. Following removal, the brains were examined grossly and then underwent parafinization. Serial sections (10 μm) were obtained on a vibratome every 250 μm and stained with hematoxylin and eosin. The sections were further examined under light microscopy for qualitative characterization of the damage. In addition, we calculated the volumes of the contusion and the hippocampi bilaterally to calculate the ipsilateral (injure)/contralateral (uninjured) hippocampal ratio. In the serial sections, the margins of both the contusion and the hippocampi were outlined separately by a blinded observer using image analysis (Imaging Research, Saint Catherines, Ontario) in all of the cut sections for each of the injured animals. Contusion and hippocampal areas were then calculated and the contusion and hippocampal volumes estimated according to the formula based on the Cavalieri Method:
EA is the summed areas of the contusion and hippocampus and t nom is the nominal section thickness of 250 μm. The contusion and hippocampal volumes is estimated as the summed volumes of each section and expressed in cubic millimeters.
Statistical analysis
All data are expressed as group mean ± standard error of the mean (SEM). Comparisons between groups for functional testing were made using a separate repeated measures analysis of variance (ANOVA; injury level × PID). If a significant effect was found in the ANOVA, individual group comparisons across days were made with Tukey’s post hoc tests. Analyses were performed using SPSS statistical software. For the histologic analysis, comparisons between groups were also made using a repeated ANOVA (injury level × PID) and Chi-square for intergroup comparisons. Statistical significance was obtained with p values of <0.05.
Results
Neurologic recovery and mortality
At all injury severities in both age groups, CCI for the deflections reported produced infrequent acute mortality. There was delayed mortality in four rats (three and one in PND 7 and 17, respectively). All surviving rats were capable of maintaining spontaneous respiration during and immediately following CCI, with only occasional transient apnea (2–3 s). With the termination of anesthesia, all surviving animals regained spontaneous movement within 2 min and were ambulating within 7 min. When returned to their litters, injured animals were initially solitary and docile. Within 2 h, the injured pups joined their littermates and initiated nursing. No gross motor dysfunction was observed prior to MWM testing. Each age at injury model is reported separately for MWM performance and histologic analysis.
CCI model in immature PND 7 rats
Morris water maze performance following CCI: PND 7
During the 5 days of MWM testing, the 1.75- and 2.0-mm CCI groups demonstrated longer swim latencies than the sham (p < 0.05) (Fig. 1a). Visual probe evaluations at PID 16–17 showed no difference between the sham group and the 1.5 mm. In contrast, there were significant differences between sham vs. the 1.75- and 2.0-mm CCI groups with visual probe evaluation. Thus, nonspecific deficits likely contribute, at least in part, to the overall poor performance of these more severely injured groups. In the probe trial, comparing time spent in the “target quadrant” vs. “out of quadrant” for each group showed that sham and 1.5 mm CCI injured animals spent significantly more time in the “target quadrant” (32.4 ± 1.96 and 28.4 ± 3.09 s, respectively) than “out of quadrant” when compared with the 1.75- and 2.0-mm CCI-injured subjects (17.1 ± 1.01 and 15.8 ± 1.47 s, respectively; p < 0.05). No statistical difference was noted between the sham and 1.5-mm CCI-injured groups or between the 1.75- and 2.0-mm CCI-injured groups.

Morris water maze functional outcome following CCI injury in a PND 7 and b 17. Note graded performance based on injury severity for each age at injury model
Histology: gross and light microscopy (PND 7)
While all the sham brains across all time points appeared grossly normal, gross examination of the injured PND 7 brains revealed a graded pathologic response to CCI. At 24 h after injury, at each deflection level of CCI, the injured brains appeared edematous and swollen with substantial subarachnoid and peri-contusional hemorrhage (Fig. 2). Histologic examination of the PND 7 injured rats confirmed the cortical edema in all CCI injured brains acutely, along with intraparenchymal (cortical) and subarachnoid hemorrhage on PID 1 (Table 1). For 1.75 and 2.0 mm, the CCI-injured brains contained large cavities at the site of impact which by PID 28, had increased in size from PID 1 (Fig. 3). Histologically, the cavitation of the cortex findings was evident at PID 28 in all CCI injured brains. This contusion/cavitation chronically involved only the cortex at all the injury levels and more extensively involved the hippocampus at the 2.0-mm injury level. The hippocampi of the 1.5-mm CCI-injured brains appeared grossly normal. Overall, atrophy and retraction of the hippocampus was noted in the more severely injured (1.75 and 2.0 mm). Ventriculomegaly was noted, to some degree, in all the 1.75- and 2.0-mm injured brains at PID 28. In contrast, for the 1.5-mm deflection, only two of ten CCI injured brains showed evidence of ventriculomegaly at PID 28. (Fig. 4a, b)

At 24 h after CCI injury, in both PND 7 and 17, all of the different severities resulted in the injured brains appearing edematous and swollen with substantial subarachnoid and peri-contusional hemorrhage for both age at injury models. Additionally, the size of the contused portion of brain increased in each age at injury based on the level of deflection severity

Representative comparison of contusion size from PID 1 to 28 following CCI in PND 7 (1.75 mm deflection) (top) and PND 17 (2.0 mm deflection) (bottom). For PND 7, the 1.75 mm CCI created a cavity at the site of impact by PID 1 which by PID 28, had increased in size. For PND 17, the 2.0 mm CCI also created a large cavity at the site of impact though by PID 28, showed only a small change from the size of the injured area at PID 1

Representative microscopic histologic findings of CCI in immature rats for a PND 7 and b PND 17 at PID 28. Note that there is increasing cortical damage with increasing injury severity. Also notable is the minimal involvement of the underlying hippocampus and diencephalic structures except at the highest injury deflections for both age at injury levels
Contusion volume, hippocampal volume, and L/R ratio (PND 7)
For PND 7, following CCI, the mean contusion volume on PID 1, increased with increased injury deflection/severity (1.5 mm = 2.73 ± 0.66 mm3; 1.75 = 3.57 ± 0.37 mm3; and 2.0 mm = 5.15 ± 1.05 mm3; p = 0.06). By PID 28, the contusion volume approximately doubled from PID 1 regardless of the level of severity and significantly differed from PID 1 at the 1.5 mm = 6.76 ± 1.34 mm3 (p = 0.02) and 1.75 mm = 7.08 ± 1.16 mm3 (p = 0.006) deflections but not the 2.0 mm = 10.96 ± 2.94 mm3 (p = 0.06)). The differences between injury levels at PID 28 were not significant (p = 0.08) (Fig. 5a).

Contusion volume comparisons of the different ages at injury for both level of injury and post-injury day (PID). For a PND 7, there was no difference between injury severity levels but there were differences in contusion volume between PID 1 and 28 for the lesser injury severities. For b PND 17, there was a significant increase in contusion volume due to injury severity only on PID 28 but also from PID 1 and 28 for the 1.5- and 2.5-mm deflections. *p < 0.05 for injury level and ** p < 0.05 for PID
Following CCI, the ipsilateral hippocampal volume and injured/non injured hippocampal ratio did not differ between injury levels (hippocampal volume—1.5 mm = 2.02 ± 0.2 mm3; 1.75 = 2.02 ± 0.3 mm3; and 2.0 mm = 1.28 ± 0.3 mm3) and (L/R hippocampal ratio—1.5 mm = 0.65 ± 0.08; 1.75 = 0.66 ± 0.09; and 2.0 mm = 0.42 ± 0.08). By PID 28, the hippocampal volumes decreased for all of the levels of severity except for the higher injury levels (hippocampal volume—1.5 mm = 1.83 ± 0.44 mm3; 1.75 = 1.33 ± 0.24 mm3; and 2.0 mm = 1.47 ± 0.45 mm3) though the differences between injury levels and over time were not significant. Hippocampal ratio also did not differ between injury levels at PID 28 except in the 1.75-mm injured animals (L/R hippocampal ratio—1.5 mm = 0.57 ± 0.13 mm3 (p = 0.6); 1.75 = 0.36 ± 0.06 mm3 (p = 0.02); and 2.0 mm = 0.44 ± 0.09 mm3 (p = 0.8)).
CCI model in immature PND 17 rats
Morris water maze performance following CCI: PND 17
For the PND 17 CCI-injured animals, the three CCI-injured groups displayed significantly longer escape latencies compared with sham (p < 0.05) (Fig. 1b). The 1.5-mm group had significantly shorter swim latencies than the 2.0- and 2.5-mm groups, but there was no difference between the 2.0- and 2.5-mm injury groups. In the probe trial, the sham and 1.5-mm CCI-injured animals spent significantly more time in the “target quadrant” (32.4 ± 1.96 and 28.4 ± 3.09 s, respectively) than “out of quadrant” when compared with the 2.0- and 2.5-mm CCI-injured subjects (17.1 ± 1.01 and 15.8 ± 1.47 s, respectively; p < 0.05). No statistical difference was noted between the sham and the 1.5-mm CCI-injured groups or between the 2.0- and 2.5-mm CCI-injured groups. Visible platform evaluations at PID 16–17 did not show a difference between sham and 1.5 mm CCI despite impaired escape latency at the 1.5-mm injury level, indicative of no nonspecific motivational or visual deficits at this injury level. In contrast, there were significant differences between sham and the 2.0 and 2.5 mm CCI groups with visual probe evaluation indicating in CCI that nonspecific deficits likely contribute, at least in part, to the overall poor performance of these more severely injured groups.
Histology: gross and light microscopy (PND 17)
On gross examination for the PND 17, the injured brains appeared pathologic at all time points and for all deflections though again in a graded response to the CCI. The brains appeared edematous and swollen with substantial subarachnoid and peri-contusional hemorrhage at PID 1 (Fig. 2). All those at the 1.5- and 2.0-mm deflections resulted in cavities at the site of impact with loss of cortex in the region of impact though seemingly larger for the 1.5-mm injury at PID 1 as compared with PID 28 with gliotic maturation and contraction of the cortical cavity grossly. The exception was the 2.5-mm deflection that seemed to have an increase in the size of the contusion cavity on PID 28 as compared with PID 1 (Fig. 3).
Histologically, on PID 1, the 1.5-mm deflection resulted in edema and only minimal intraparenchymal and subarachnoid hemorrhage and minimal contusion. Cortical necrosis and cavity formation approaching but not involving the underlying hippocampus at the 2.0- and 2.5-mm injury levels. While the hippocampi of the 1.5-mm CCI-injured brains appeared grossly normal, atrophy and retraction of the hippocampus was noted in the more severely injured (2.0 and 2.5 mm deflections) (Fig. 4b). Similarly, ventriculomegaly was noted, to some degree, in all the 2.0- and 2.5-mm CCI-injured brains at PID 28 though at the 1.5-mm deflection, only one of ten injured brains showed evidence of ventriculomegaly at PID 28.
Contusion volume, hippocampal volume, and L/R ratio (PND 17)
For PND 17, following CCI, contusion volume did not differ with increased injury severity on PID 1 (1.5 mm = 8.13 ± 1.48 mm3; 2.0 = 8.59 ± 1.19 mm3; and 2.5 mm = 8.6 ± 1.4 mm3; p = 0.95) but by PID 28, there was a significant difference in contusion volume across injury severities (1.5 mm = 4.32 ± 0.7 mm3; 2.0 mm = 8.68 ± 1.37 mm3; and 2.5 mm = 14.67 ± 1.81 mm3; p < 0.0001) (Fig. 5b). For the 1.5- and the 2.5-mm injury levels (p < 0.05), there were significant differences between PID 1 and 28 though not the 2.0-mm deflection (p = 0.95). Interestingly, the contusion volume was significantly smaller in the 1.5-mm deflection at PID 28 as compared with PID 1 likely secondary to the contraction of the gliotic scar.
In the injured hippocampus, on PID 1, hippocampal volume and hippocampal ratio did not differ with increasing severity (hippocampal volume—1.5 mm = 2.36 ± 0.2 mm3; 2.0 = 2.54 ± 0.23 mm3; and 2.5 mm = 2.53 ± 0.29 mm3; p = 0.97) and (L/R hippocampal ratio—1.5 mm = 0.82 ± 0.05; 2.0 = 0.81 ± 0.08; and 2.5 mm = 0.80 ± 0.06; p = 0.85) likely indicating no direct injury to the hippocampus. By PID 28, the hippocampal volumes significantly decreased as compared with PID 1 in the more severe levels of injury (hippocampal volume—1.5 mm = 2.45 ± 0.26 mm3 (p = 0.8); 2.0 = 1.47 ± 0.25 mm3 (p = 0.01); and 2.5 mm = 1.62 ± 0.23 mm3 (p = 0.02)) as well as across the three different injury levels (p = 0.01). Hippocampal ratio also decreased in all injury levels though again not at the 1.5-mm deflection (L/R hippocampal ratio—1.5 mm = 0.78 ± 0.06 (p = 0.6); 2.0 = 0.46 ± 0.07 (p = 0.004); and 2.5 mm = 0.47 ± 0.07 (p = 0.006)) For PND 17 as a group, there was a significant difference between injury severity and PID (p = 0.001).
Discussion
TBI clinically does not result in a single characteristic type of injury due to the differences in the primary mechanism, the direction and acceleration of forces on the brain, the severity, the second insults, and then the concomitant secondary response. While there have been multiple models of experimental TBI to help better understand the response to TBI, age at injury has not been as well characterized compared with mature adult brain injury. The characterization and further development of new models in the immature are needed to investigate the unique pathophysiology of the developing brain following injury and the development of potentially novel therapeutic interventions. Based on our clinical observations our focus in developing models of injury in the immature is that any new model results in a functional deficit. For this study, using CCI, we have further characterized a contusion model of TBI with graded severity of injury at PND 17 in the immature rat and developed a new model of experimental, contusive TBI in the PND 7. The injuries produced a graded injury with increasing cortical deflection that resulted in: (1) significant MWM performance deficits of spatial memory, (2) acute and chronic histopathologic lesions, and (3) chronic changes in both contusion and hippocampal volumes and ratios in the two models of cortical injury in immature rats (PND 7 and 17). The PND 7 model resulted in a graded response for the acute injury at the acute and more chronic time points with regard to contusion volume and hippocampal damage. In the PND 17 model, the differences between injury severities were not histologically obvious at PID 1 but were more pronounced at PID 28.
CCI in the immature rat
As mentioned earlier, there have been a number of other developmental pediatric TBI models that have attempted to better understand the impact of experimental TBI in the developing brain. For the most part, to date in experimental CCI, studies have focused on important potential therapeutic avenues of special interest with regard to the developing brain and its vulnerability, such as oxidative stress [11, 21] alternative fuels, particularly ketones [54], mitochondrial failure [60], and proteomic response [33, 35] without defining the histopathologic nor functional response of the developing brain to injury. In this study, we sought to more definitively characterize CCI in two different ages at injury, not so much to compare effect of injury between the two ages but rather to define the unique response through a model of experimental TBI at each age at injury.
Effect of CCI on MWM performance
Immature rats have been shown to be capable of learning the MWM task [38, 62] that assesses hippocampal mediated visuo-spatial memory and place navigation attaining adult levels of proficiency between PND 21–23 [9]. It is unclear though whether MWM assesses spatial memory acquisition in developing animals [61]. Similarly, it is a challenge to clinically identify in children a accurate and sufficient “outcome measure” to assess cognitive function following TBI. Still, the MWM is a valuable adjunct for identifying a performance deficit that has likely subcortical origins and presently provides a well described, and studied endpoint for measure following experimental TBI. The literature regarding MWM performance after TBI in immature rats has been growing [3, 4, 6], and to our knowledge, no data exist regarding MWM performance after CCI in immature rats as well as MWM performance following injury in PND 7. Consistent with previously described maturational timelines [9], sham rats in this study beginning MWM testing on PND 27 were capable of learning the task. The initial MWM testing of the two models took place at PND 18 for the younger at injury model and PND 28 for the older model. Swim speeds did not markedly differ from that of adult and from what we had previously reported for these age at testing animals in these models [3, 6].
MWM impairments have been shown after both hippocampal [50, 65] and cortical [12] lesions in adult rats, leading investigators to relate the cognitive deficits to histopathologic damage. MWM performance and outcome following experimental TBI has been variable when utilized in the immature rat. After fluid-percussion injury, PND 17 rats had no escape latency deficits but did exhibit poor probe trial performance as compared with shams [55]. In contrast, PND 17 animals undergoing diffuse impact acceleration injury showed significantly prolonged escape latencies after a 150-g/2 m injury that were sustained through the third post-injury month [3]. In the present study, significant MWM acquisition deficits were found following 1.75 and 2.0 mm in the PND 7 model and the 2.0- and 2.5-mm deflections in the PND 17 model. Interestingly, a 1.5-mm level of injury resulted in MWM acquisition deficits in the PND 17, but not the PND 7 group, suggesting a potential age-dependent injury threshold.
The minimal numbers of studies examining MWM deficits after TBI in the immature rat have failed to correlate acute cognitive deficits in MWM performance to histopathology [4, 55] though it has been suggested that insult and deafferentation may contribute to MWM performance deficits [57]. Mechanisms other than tissue damage have been attributed to MWM deficits including cortical cholinergic system dysfunction [12] and septohippocampal cholinergic systems [24] but to date this has not been examined in the immature rat. Additionally, it is possible that sensorimotor-based exploratory behavior is suppressed after CCI in the immature rat as sensory attention and integration functions begin to appear at the time that the rats were injured in this study [59].
Histopathologic injury
In contrast to previously described models of immature TBI, little acute or delayed mortality were seen in either age or severity injury level. Mortality rates of 27–100 % using fluid percussion [56], 8–9 % using focal weight drop [46], and up to 83 % using diffuse weight drop/impact acceleration [6] have been previously described. By definition, diffuse weight drop models of TBI have more widespread neuropathologic effects, likely including significant brainstem injury or physiologic interruption. Fluid percussion, although a more focal model of TBI produces diffuse central nervous system (CNS) pathology including brainstem hemorrhage [49] and severe brainstem strain [63]. Unlike other models of immature TBI, CCI produces a lesion that is primarily focal in nature, leaving the brainstem intact. By minimizing the injury to critical central respiratory drive centers in the CNS, the CCI model of TBI applied to the PND 7 and 17 was able to produce a cortical injury with a 0 % primary mortality rate in our hands. In addition, by delivering anesthesia via inhalation, the need for tracheal intubation, as in the impact acceleration model [6], is eliminated. In our experience, intubation of PND 17 rat pups is associated with post-intubation tracheal spasm and edema and occasional intubation-related death [3].
In this model of TBI in the immature rat using CCI produced focal cortical tissue loss in a graded manner, the magnitude correlating with depth of brain deflection. At the more severe injury levels in the PND 17 (2.0 and 2.5 mm), the initially contused cortex evolved into a cortical cavity that extended to, but did not involve directly, the underlying hippocampus. Interestingly, hippocampal asymmetry was noted in both age at injury models over the study period at PID 1 in the most severe injury deflection (PND 7, 2.0 mm and PND 17, 2.5 mm deflection) and most of the injuries at PID 28 (PND 7, 1.75 and 2.0 mm and PND 17, 2.0 and 2.5 mm deflections) indicating some level of “transmitted” injury and potentially allowing for further study of post-injury hippocampal pathology that may be contributory to the MWM deficits noted in these models. The ability to produce a graded contusion is a unique feature of the CCI model, and comparable histopathology has been observed in adult CCI models [18, 44]. These age at injury models using CCI have a number of histopathological advantages. By manipulating brain deflection and impactor tip, the resultant injury was able to be reliably and consistently altered to produce the desired degree of histopathologic injury of contusion and hippocampal asymmetry (without direct damage) correlative with MWM functional outcome.
Lastly, CCI might produce a more diffuse injury in addition to the focal contusion with the tissue deformation from the propagated energy within in the closed skull not only to the hippocampus but throughout the cerebrum and infratentorial may also be contributory to the diffuse findings in these two models [28]. As a result, the analysis of the extent of ipsilateral and contralateral neurodegeneration provides a more complete anatomical correlate for the cognitive and motor dysfunction seen in this paradigm and suggests that visual disturbances may are also likely to be contributory involved in the post-CCI neurological deficits observed. As well, TBI in infants is often caused by abusive head trauma. This form of TBI has not been modeled in rats and therefore, this experimental paradigm in the PND 7, which models a newborn, could also provide some insight into aspects of the pathobiology of that important condition.
Conclusions
Utilizing two different ages at injury models of immature TBI, we have demonstrated significant cortical and hippocampal histologic damage, both acute and delayed and cognitive functional deficits in both PND 7 and 17 individually after CCI. We have shown a graded response for histologic damage with increasing deflection depth. Since the immature brain differs from the adult and given the impact of TBI on the pediatric population, it is critical that TBI in the immature brain be further explored. These experimental animal models of TBI in the immature brain should provide a template for further investigations into the mechanisms and treatments of immature brain injury.
References
Adelson PD (1999) Animal models of traumatic brain injury in the immature: a review. Exp Toxicol Pathol 51(2):130–136
Adelson PD, Bratton SL, Carney NA, Chesnut RM, du Coudray HE, Goldstein B, Kochanek PM, Miller HC, Partington MD, Selden NR, Warden CR, Wright DW (2003) Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Pediatr Crit Care Med 4(3 Suppl):S1–S75
Adelson PD, Dixon CE, Kochanek PM (2000) Long-term dysfunction following diffuse traumatic brain injury in the immature rat. J Neurotrauma 17(4):273–282
Adelson PD, Dixon CE, Robichaud P, Kochanek PM (1997) Motor and cognitive functional deficits following diffuse traumatic brain injury in the immature rat. J Neurotrauma 14(2):99–108
Adelson PD, Jenkins LW, Hamilton RL, Robichaud P, Tran MP, Kochanek PM (2001) Histopathologic response of the immature rat to diffuse traumatic brain injury. J Neurotrauma 18(10):967–976
Adelson PD, Robichaud P, Hamilton RL, Kochanek PM (1996) A model of diffuse traumatic brain injury in the immature rat. J Neurosurg 85(5):877–884
Adelson PD, Whalen MJ, Kochanek PM, Robichaud P, Carlos TM (1998) Blood brain barrier permeability and acute inflammation in two models of traumatic brain injury in the immature rat: a preliminary report. Acta Neurochir Suppl 71:104–106
Armstead WM (1998) Brain injury impairs prostaglandin cerebrovasodilation. J Neurotrauma 15(9):721–729
Bachevalier J, Beauregard M (1993) Maturation of medial temporal lobe memory functions in rodents, monkeys, and humans. Hippocampus 3(Spec No):191–201
Bayir H, Kochanek PM, Kagan VE (2006) Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci 28(4–5):420–431
Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE (2007) Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol 62(2):154–169
Berger-Sweeney J, Heckers S, Mesulam MM, Wiley RG, Lappi DA, Sharma M (1994) Differential effects on spatial navigation of immunotoxin-induced cholinergic lesions of the medial septal area and nucleus basalis magnocellularis. J Neurosci: Off J Soc Neurosci 14(7):4507–4519
Berger MS, Pitts LH, Lovely M, Edwards MS, Bartkowski HM (1985) Outcome from severe head injury in children and adolescents. J Neurosurg 62(2):194–199
Bullock MR, Chesnut R, Ghajar J, Gordon D, Hartl R, Newell DW, Servadei F, Walters BC, Wilberger JE (2006) Guidelines for the surgical management of traumatic brain injury. Neurosurgery 58(3 Suppl):S2-iv
Clark RS, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH (1997) Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci 17(23):9172–9182
Clark RS, Kochanek PM, Dixon CE, Chen M, Marion DW, Heineman S, DeKosky ST, Graham SH (1997) Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma 14(4):179–189
Clark RS, Kochanek PM, Schwarz MA, Schiding JK, Turner DS, Chen M, Carlos TM, Watkins SC (1996) Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr Res 39(5):784–790
Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39(3):253–262
Dixon CE, Markgraf CG, Angileri F, Pike BR, Wolfson B, Newcomb JK, Bismar MM, Blanco AJ, Clifton GL, Hayes RL (1998) Protective effects of moderate hypothermia on behavioral deficits but not necrotic cavitation following cortical impact injury in the rat. J Neurotrauma 15(2):95–103
Duhaime AC, Margulies SS, Durham SR, O’Rourke MM, Golden JA, Marwaha S, Raghupathi R (2000) Maturation-dependent response of the piglet brain to scaled cortical impact. J Neurosurg 93(3):455–462
Fan P, Yamauchi T, Noble LJ, Ferriero DM (2003) Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma 20(5):437–445
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG (1981) Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res 211(1):67–77
Fink EL, Alexander H, Marco CD, Dixon CE, Kochanek PM, Jenkins LW, Lai Y, Donovan HA, Hickey RW, Clark RS (2004) Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatric Crit Care Med 5(2):139–144
Frielingsdorf H, Thal LJ, Pizzo DP (2006) The septohippocampal cholinergic system and spatial working memory in the Morris water maze. Behav Brain Res 168(1):37–46
Gao WM, Chadha MS, Kline AE, Clark RS, Kochanek PM, Dixon CE, Jenkins LW (2006) Immunohistochemical analysis of histone H3 acetylation and methylation–evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res 1070(1):31–34
Graham DI, Ford I, Adams JH, Doyle D, Lawrence AE, McLellan DR, Ng HK (1989) Fatal head injury in children. J Clin Pathol 42(1):18–22
Grundl PD, Biagas KV, Kochanek PM, Schiding JK, Barmada MA, Nemoto EM (1994) Early cerebrovascular response to head injury in immature and mature rats. J Neurotrauma 11(2):135–148
Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW (2005) Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma 22(2):252–265
Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG, Hayes RL (1992) Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma 9(1):11–20
Huh JW, Raghupathi R (2007) Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat. J Neurotrauma 24(9):1460–1474
Huh JW, Widing AG, Raghupathi R (2007) Basic science; repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: a preliminary report. J Neurotrauma 24(1):15–27
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283(5398):70–74
Jenkins LW, Peters GW, Dixon CE, Zhang X, Clark RS, Skinner JC, Marion DW, Adelson PD, Kochanek PM (2002) Conventional and functional proteomics using large format two-dimensional gel electrophoresis 24 hours after controlled cortical impact in postnatal day 17 rats. J Neurotrauma 19(6):715–740
Johnson DL, Krishnamurthy S (1998) Severe pediatric head injury: myth, magic, and actual fact. Pediatr Neurosurg 28(4):167–172
Kochanek AR, Kline AE, Gao WM, Chadha M, Lai Y, Clark RS, Dixon CE, Jenkins LW (2006) Gel-based hippocampal proteomic analysis 2 weeks following traumatic brain injury to immature rats using controlled cortical impact. Dev Neurosci 28(4–5):410–419
Kochanek PM (2006) Pediatric traumatic brain injury: quo vadis? Dev Neurosci 28(4–5):244–255
Kochanek PM, Carney N, Adelson PD, Ashwal S, Bell MJ, Bratton S, Carson S, Chesnut RM, Ghajar J, Goldstein B, Grant GA, Kissoon N, Peterson K, Selden NR, Tasker RC, Tong KA, Vavilala MS, Wainwright MS, Warden CR (2012) Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents—second edition. Pediatric Crit Care Med 13(Suppl 1):S1–S82
Kraemer PJ, Brown RW, Baldwin SA, Scheff SW (1996) Validation of a single-day Morris water maze procedure used to assess cognitive deficits associated with brain damage. Brain Res Bull 39(1):17–22
Kraus JF (1987) Epidemiology of head injury. In: Cooper PR (ed) Head injury. Williams and Wilkins, Baltimore, pp 1–19
Kraus JF (1995) Epidemiological features of brain injury in children: occurrence, children at risk, causes and manner of injury, severity, and outcomes. In: Broman SH, Michel ME (eds) Traumatic head injury in children. Oxford University Press, New York, pp 22–39
Kraus JF, Fife D, Conroy C (1987) Pediatric brain injuries: the nature, clinical course, and early outcomes in a defined United States’ population. Pediatrics 79(4):501–507
Levin HS, Eisenberg HM, Wigg NR, Kobayashi K (1982) Memory and intellectual ability after head injury in children and adolescents. Neurosurgery 11(5):668–673
Levin HS, Ewing-Cobbs L, Eisenberg HM (1995) Neurobehavioral outcome of pediatric closed head injury. In: Broman SH, Michel M (eds) Traumatic head injury in children. Oxford University Press, New York, pp 70–94
Lighthall JW, Dixon CE, Anderson TE (1989) Experimental models of brain injury. J Neurotrauma 6(2):83–97
Luerssen TG, Klauber MR, Marshall LF (1988) Outcome from head injury related to patient’s age. A longitudinal prospective study of adult and pediatric head injury. J Neurosurg 68(3):409–416
Mansfield RT, Schiding JK, Hamilton RL, Kochanek PM (1996) Effects of hypothermia on traumatic brain injury in immature rats. J Cereb Blood Flow Metab 16(2):244–252
McAllister TW (2011) Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci 13(3):287–300
McDonald JW, Johnston MV (1990) Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev 15(1):41–70
McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL (1989) Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience 28(1):233–244
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11(1):47–60
Moser E, Mathiesen I, Andersen P (1993) Association between brain temperature and dentate field potentials in exploring and swimming rats. Science 259(5099):1324–1326
Nehlig A, de Vasconcelos AP, Boyet S (1988) Quantitative autoradiographic measurement of local cerebral glucose utilization in freely moving rats during postnatal development. J Neurosci 8(7):2321–2333
Nehlig A, Pereira de Vasconcelos A, Boyet S (1989) Postnatal changes in local cerebral blood flow measured by the quantitative autoradiographic [14C]iodoantipyrine technique in freely moving rats. J Cereb Blood Flow Metab 9(5):579–588
Prins ML, Fujima LS, Hovda DA (2005) Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res 82(3):413–420
Prins ML, Hovda DA (1998) Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. J Neurotrauma 15(10):799–811
Prins ML, Lee SM, Cheng CL, Becker DP, Hovda DA (1996) Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Res Dev Brain Res 95(2):272–282
Prins ML, Povlishock JT, Phillips LL (2003) The effects of combined fluid percussion traumatic brain injury and unilateral entorhinal deafferentation on the juvenile rat brain. Brain Res Dev Brain Res 140(1):93–104
Raghupathi R, Huh JW (2007) Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J Neurotrauma 24(10):1596–1608
Rice JE 3rd, Vannucci RC, Brierley JB (1981) The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9(2):131–141
Robertson CL, Saraswati M, Fiskum G (2007) Mitochondrial dysfunction early after traumatic brain injury in immature rats. J Neurochem 101(5):1248–1257
Rudy JW, Stadler-Morris S, Albert P (1987) Ontogeny of spatial navigation behaviors in the rat: dissociation of “proximal”- and “distal”-cue-based behaviors. Behav Neurosci 101(1):62–73
Schenk F (1989) A homing procedure for studying spatial memory in immature and adult rodents. J Neurosci Methods 26(3):249–258
Shima K, Marmarou A (1991) Evaluation of brain-stem dysfunction following severe fluid-percussion head injury to the cat. J Neurosurg 74(2):270–277
Stevenson KL, Davis DS, Skinner JC, Tran MP, Dixon CE, Kochanek PM, Jenkins LW, Adelson PD (2000) Behavioral dysfunction in immature rats after controlled cortical impact. [Abstract]. J Neurotrauma 17(10):944
Sutherland RJ, Whishaw IQ, Regehr JC (1982) Cholinergic receptor blockade impairs spatial localization by use of distal cues in the rat. J Comp Physiol Psychol 96(4):563–573
Acknowledgments
The authors wish to thank the National Institute of Health for its support for PDA (NIH RO1 NS42298) and PMK (NIH RO1 NS 30318 and NS 38087). The authors also wish to thank Ms. Christina Casanova for her help in the manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Adelson, P.D., Fellows-Mayle, W., Kochanek, P.M. et al. Morris water maze function and histologic characterization of two age-at-injury experimental models of controlled cortical impact in the immature rat. Childs Nerv Syst 29, 43–53 (2013). https://doi.org/10.1007/s00381-012-1932-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-012-1932-4