
Abstract
Purpose
The purpose of this study was to estimate the association among the presence of subependymal nodules (SENs), subependymal giant cell tumours (SGCTs) and gene mutation in tuberous sclerosis complex (TSC) patients.
Methods
Clinical records and images of 81 TSC patients were retrospectively reviewed by two neuroradiologists in consensus. All patients were assessed for gene mutations and were categorized as TSC1 or TSC2 mutation carriers, or no-mutations-identified (NMI) patients. They underwent a brain magnetic resonance imaging (MRI) using 0.1 mmol/kg of gadobutrol. Any enhancing SEN ≥ 1 cm and placed near the foramen of Monro was considered SGCT. Two MRI follow-up exams for each patient with SGCT were evaluated to assess tumour growth using Wilcoxon and chi-squared tests.
Results
Of 81 patients, 44 (54 %) were TSC2 mutation carriers, 20 (25 %) TSC1 and 17 (21 %) NMI. Nine (11 %) had a unilateral and three (4 %) a bilateral SGCT. Fifty of 81 patients (62 %) showed at least one SEN. None of the 31 patients without SEN showed SGCTs, whilst 12 (24 %) of the 50 patients with at least one SEN showed SGCTs (p = 0.003). The association between the presence of SGCT or SEN and gene mutation was not significant (p = 0.251 and p = 0.187, respectively). At follow-up, the median SGCT diameter increased from 14 to 15 mm (p = 0.017), whilst the median SGCT volume increased from 589 to 791 mm3 (p = 0.006).
Conclusions
TSC patients with SENs are more likely to present with SGCT than those without SENs, in particular for TSC2 mutation carriers. The SGCT growth rate may be missed if based on the diameter instead of on the volume.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant multisystem disease resulting from the improper differentiation, proliferation or migration of neuroglial progenitor cells during foetal development [1–3]. Its prevalence is of 9 in 100,000 individuals [4], with clinical presentation depending on mutations in either of two genes—TSC1 (located on 9q34) or TSC2 (located on 16p13)—encoding for hamartin and tuberin, respectively [5].
Current molecular diagnostic methods permit the identification of mutations in either TSC1 or TSC2 gene in only 75–85 % of TSC patients [6]. Therefore, as many as 20 % of patients with the TSC phenotype has no mutations identified (NMI) [7].
Brain involvement may include cortical or subcortical tubers, subependymal nodules (SENs), subependymal giant cell tumour (SGCTs) and white matter abnormalities [8–10]. According to published series, the prevalence reported of SGCT in TSC patients is from 5 to 20 % [11–13]. Dabora et al. [14] studied a large sample of 224 TSC patients showing an SGCT prevalence of 11 % overall, 9 % among TSC1 mutation carriers and 12 % among TSC2 mutation carriers. They also showed a higher prevalence of SENs among TSC2 than in TSC1 mutation carriers.
In the present study, we aimed at evaluating the association among the prevalence of SENs or SGCTs and the genetic mutational status in a population of TSC patients.
Materials and methods
Study population
This study has been approved by the Institutional Review Board of our hospital and complies with the standards of the Declaration of Helsinki and current ethical guidelines.
Clinical records and images of all the 81 TSC patients referred at the radiology department of our Institution from June 2005 to June 2010 for a brain magnetic resonance imaging (MRI) examination were retrospectively reviewed by two neuroradiologists in consensus. They were 31 males and 50 females, with a mean age of 28 years (range, 3–65 years). According to clinical records, they had a median age at diagnosis of TSC of 10 months (25th percentile, 5 months; 75th percentile, 30 months). All of them had a genetic assessment for gene mutations and were categorized as having the TSC1 mutation, the TSC2 mutation or as NMI patients. All patients affected with SGCT underwent a second brain MRI as a follow-up examination.
Imaging protocol and image analysis
All examinations were performed using a 1.0-T unit (Intera 1.0 T, Philips Medical Systems, Best, the Netherlands). For each patient, MRI was performed according to the protocol described in Table 1.
An SGCT was defined as a markedly enhancing lesion larger than 1 cm in its maximal diameter and near the foramen of Monro. For each SGCT, we measured the maximal diameter in the three dimensions (D1, D2 and D3) and estimated the lesion volume applying the following ellipsoid formula:
which falls into the sphere volume formula if D 1 = D 2 = D 3. Moreover, the number, side and presence of calcifications were evaluated.
Statistical analysis
We estimated the association among the presence of SGCT, the presence of SEN and mutational status using the χ 2 test. Odds ratios and 95 % confidence intervals (CI) were calculated.
Referring to SGCTs, the distribution of the maximal diameter and of the volume was calculated at the first and at the second examination. The difference in between was estimated using the Wilcoxon signed-rank test for paired data. We also calculated the percentage of patients with a 20 % increase in both diameter and volume.
All analyses were performed using SPSS Statistics v. 17 (SPSS, Inc., v.17, Chicago, IL), and p values <0.05 were considered statistically significant.
Results
Distributions
Out of 81 patients, 17 (21 %) were NMI, 20 (25 %) had TSC1 mutation, and 44 (54 %) had TSC2 mutation. Nine patients (11 %) had a unilateral and three patients (4 %) had bilateral tumours, for a total of 15 SGCTs. Fifty (62 %) showed at least one SEN.
Correlations
At the bivariate correlation analysis, the presence of either SGCT or SEN was not associated with the genetic mutation (p = 0.251 and p = 0.187, respectively). However, a trend for a higher prevalence of SGCTs among TSC2 patients (9/44, 20 %) with respect to the subset of NMI and TSC1 patients (3/37, 8 %) was observed: the odds ratio was 2.9 (CI = 0.7–11.7). Similarly, there was a higher prevalence of patients with at least one SEN among TSC2 patients (31/44, 70 %) with respect to the subset of NMI and TSC1 patients (19/37, 51 %): the odds ratio was 2.3 (CI = 0.9–5.6). All these distributions are shown in Table 2.
In our population, a significant association between the detection of further SEN in patients carrying SGCT was observed: in fact, none of the 31 patients without SEN showed an SGCT, whilst 12 (24 %) of the 50 patients with at least one SEN showed an SGCT (p = 0.003). The odds ratio was not calculable due to the presence of a zero.
SGCT patients
Considering only the 12 SGCT patients, the median age at the first MRI performed at our institution was 22 years (25th percentile, 18 years; 75th percentile, 33 years), whilst the mean age of SGCT diagnosis according to clinical records was 17 years. Calcifications were observed in 11 out of 15 tumours, whilst ventriculomegaly was present in seven (58 %) patients, only one of them being symptomatic and needing neurosurgery. One patient, operated in an early age, came to our attention for recurrence.
From the first to the second MRI examination, a median of 2.5 years passed (25th percentile, 2.1 years; 75th percentile, 3.6 years). Between the two examinations, the median SGCT diameter significantly increased from 14 to 15 mm (p = 0.017), whilst the tumour volume significantly increased from 589 to 791 mm3 (p = 0.006). Evolutions of tumour diameter and volume are shown in Fig. 1. Out of the 15 SGCTs, only one (7 %) showed an increase larger than 20 % in diameter, whilst eight (53 %) showed an increase of more than 20 % in volume.

Graphs showing the temporal evolution of SGCTs in terms of diameter (a) and volume (b). Out of 15 SGCTs, only one (7 %) showed an increase larger than 20 % in diameter, whilst eight (53 %) showed an increase of more than 20 % in volume. Note that the majority of patients lie between 10 and 35 years of age and that the three older patients had a stable disease
Discussion
In this study, we evaluated the association among the presence of SEN and that of SGCT in a population of TSC patients. Moreover, we estimated the impact of a gene mutational status on these prevalence rates. The most important finding of this study is the significantly higher prevalence of SGCT among patients with at least one SEN. In fact, none of the 31 patients without SEN showed an SGCT, whilst 12 (24 %) of the 50 patients with at least one SEN showed an SGCT. In this regard, we should note that SGCTs usually correspond to growing SENs and that, cytologically, there is no difference between them [15, 16]. In fact, both lesions are of mixed glioneuronal lineage and contain giant cells [16, 17]. Transformation of a SEN into an SGCT is usually a gradual process, of which the highest rate is in the first two decades of life [16]. SEN growth peaks at puberty and stops by the end of the third decade [18]. Moreover, a SEN presenting larger than 5 mm in diameter, located at the peri-Monro region, with incomplete calcification and with a marked contrast enhancement at MRI is more likely to transform into an SGCT [19, 20].
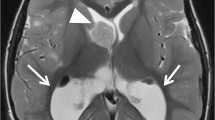
This plays in favour of a temporal correlation between SEN and SGCT. Our results show a “spatial correlation”: SENs located over lateral ventricles were significantly associated with SGCT. In fact, we may reformulate our results saying that all of the 12 patients (100 %) with an SGCT showed at least one further SEN, whilst this percentage was 51 % (35/69) among those without SGCT (p = 0.001). This result may be thought as expected in that it mostly applies to TSC2 patients who are most likely to develop SENs [14]. In this regards, our results agree with those found by Dabora et al., with about 70 % of the TSC2 patients presenting with at least a SEN. Moreover, in this population, about 20 % of patients had an SGCT. In practice, TSC2 patients are more likely to present with SENs and to develop a SGCT (Fig. 2).

Twenty-four-year-old woman affected with tuberous sclerosis complex carrying the TSC2 gene mutation. a, b Coronal and axial contrast-enhanced T1-weighted images, respectively, demonstrating a 16-mm enhancing lesion near the right foramen of Monro (arrowhead) corresponding to a lesion volume of 2,144 mm3. The tumour was associated with multiple subependymal nodules, as indicated by arrowheads in axial and T2-weighted (d) and contrast-enhanced, T1-weighted (c) images
The radiological diagnostic criteria of SGCT in TSC patients vary substantially between studies [21]. A shared consensus about imaging signs has not been reached. The definition of SGCT in the literature varies from any enhancing SEN in the brain of TSC patients [22] to only markedly enhancing lesions that are located near the foramen of Monro [23]. The diagnostic performance of these radiological criteria could be estimated against histopathology. However, few patients undergo surgery; thus, a real reference standard is scarcely available. In order to overcome this limitation and to obtain an estimation of the difference of prevalence between the radiological criteria and histopathology, Adriaensen et al. performed a meta-analysis [24]. They obtained a pooled prevalence of SGCT of 16 % using radiological criteria and 9 % using histopathology. Based on these results, we can calculate a false-positive rate of the radiological criteria of 16–9 % = 7 % and a positive predictive value of 56 % [24]. This means that on a sample of TSC patients fulfilling the radiological criteria for SGCT, only 56 % actually have an SGCT. Applying this percentage to our sample, we may speculate that only 8 out of the 15 observed SGCTs could be histopathologically confirmed. As a matter of fact, in our series, we found only 8 out of 15 radiologically determined SGCTs with a volume increase of at least 20 % (Fig. 1). Even with limitations, we may speculate that these eight lesions are more likely to be real SGCTs according to the aforementioned positive predictive value. Moreover, these data partially demonstrate that the tumour growth should be assessed looking at the volume increase instead of the diameter increase (Fig. 3).

Two brain magnetic resonance images obtained in a tuberous sclerosis complex patients carrying the TSC1 gene mutation. Image in (a) was acquired when the patient was 10 years old, whilst (b) shows the corresponding image obtained 3 years later. A subependymal giant cell tumour was clearly visible (arrowhead), with an increase in maximum diameter from 21 to 25 mm corresponding to an increase in volume from 2,813 to 4,329 mm3. Furthermore, the lesion morphology slightly changed over time
The above considerations may help clinicians in the management of patients with an SGCT, in particular those with a high growth rate. Some authors suggest preemptive neurosurgical intervention, even though it is not known how many radiologically diagnosed SGCTs really grow and cause clinical problems [20]. It is still an important debate since some clinicians are appropriately intervening and electively removing lesions that would have progressed to cause significant problems. For example, de Ribaupierre et al. [20] demonstrated that the elective resection in an asymptomatic stage reduces the risk of possible complications. However, it is not possible to exclude that some patients underwent unnecessary removal of lesions which would never have caused clinical problems. As it is for other imaging issues, the risk of overdiagnosis and overtreatment should be considered.
The natural history of SGCT has not been completely elucidated. Recommendations issued by the NIH Consensus Conference [25] suggest that all TSC patients should undergo brain MRI every 1–3 years up to 21 years of age. Shortening of the monitoring interval is suggested in case of lesion progression [25, 26]. However, subsequent published articles recommend even a more frequent monitoring [16], or not recommend screening at all [22].
Conclusions
In conclusion, TSC patients with SENs are more likely to present with an SGCT, in particular those carrying the TSC2 gene mutation. Moreover, the growth rate of these lesions should be assessed considering the volume instead of the diameter increase.
References
Grajkowska W, Kotulska K, Jurkiewicz E, Matyja E (2010) Brain lesions in tuberous sclerosis complex. Review. Folia Neuropathol 48:139–149
Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, Hukee MJ et al (1995) Tuber and subependymal giant cell astrocytoma associated with tuberous sclerosis: an immunohistochemical, ultrastructural, and immunoelectron and microscopic study. Acta Neuropathol 90:387–399
Scheithauer BW (1992) The neuropathology of tuberous sclerosis. J Dermatol 19:897–903
O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN (1998) Prevalence of tuberous sclerosis estimated by capture–recapture analysis. Lancet 351:1490
Crino PB (2004) Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol 19:716–725
Jozwiak J, Jozwiak S, Wlodarski P (2008) Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol 9:73–79
Kaczorowska M, Jurkiewicz E, Domańska-Pakieła D, Syczewska M, Lojszczyk B, Chmielewski D et al (2011) Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia 52:22–27
Moavero R, Pinci M, Bombardieri R, Curatolo P (2011) The management of subependymal giant cell tumors in tuberous sclerosis: a clinician’s perspective. Childs Nerv Syst 27:1203–1210
Houser OW, Gomez MR (1992) CT and MR imaging of intracranial tuberous sclerosis. J Dermatol 19:904–908
Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355:1345–1356
Cuccia V, Zuccaro G, Sosa F, Monges J, Lubienieky F, Taratuto AL (2003) Subependymal giant cell astrocytoma in children with tuberous sclerosis. Childs Nerv Syst 19:232–243
Bollo RJ, Berliner JL, Fischer I, Miles DK, Thiele EA, Zagzag D et al (2009) Extraventricular subependymal giant cell tumor in a child with tuberous sclerosis complex. J Neurosurg Pediatr 4:85–90
Shepherd CW, Gomez MR, Lie JT, Crowson CS (1991) Causes of death in patients with tuberous sclerosis. Mayo Clin Proc 66:792–796
Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J et al (2001) Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 68:64–80
Fujiwara S, Takaki T, Hikita T, Nishio S (1989) Subependymal giant-cell astrocytoma associated with tuberous sclerosis. Do subependymal nodules grow? Childs Nerv Syst 5:43–44
Goh S, Butler W, Thiele EA (2004) Subependymal giant cell tumors in tuberous sclerosis complex. Neurology 63:1457–1461
Shepherd CW, Scheithauer BW, Gomez MR, Altermatt HJ, Katzmann JA (1991) Subependymal giant cell astrocytoma: a clinical, pathological, and flow cytometric study. Neurosurgery 28:864–868
Torres VE, King BF, McKusick MA, Delalande O, Dulac O, Chiron C (2001) Update on tuberous sclerosis complex. Contrib Nephrol 136:33–49
Nabbout R, Santos M, Rolland Y, Delalande O, Dulac O, Chiron C (1999) Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psychiatr 66:370–375
de Ribaupierre S, Dorfmüller G, Bulteau C, Fohlen M, Pinard J-M, Chiron C et al (2007) Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery 60:83–89
Adriaensen MEAPM, Schaefer-Prokop CM, Stijnen T, Duyndam DAC, Zonnenberg BA, Prokop M (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16:691–696
O’Callaghan FJK, Martyn CN, Renowden S, Noakes M, Presdee D, Osborne JP (2008) Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: a population-based study. Arch Dis Child 93:751–754
Koeller KK, Sandberg GD (2002) From the archives of the AFIP. Cerebral intraventricular neoplasms: radiologic–pathologic correlation. Radiographics 22:1473–1505
Sardanelli F, Di Leo G (2009) Biostatistics for radiologists: planning, performing, and writing a radiologic study. Springer, Milan
Hyman MH, Whittemore VH (2000) National Institutes of Health consensus conference: tuberous sclerosis complex. Arch Neurol 57:662–665
Clarke MJ, Foy AB, Wetjen N, Raffel C (2006) Imaging characteristics and growth of subependymal giant cell astrocytomas. Neurosurg Focus 20:E5
Conflict of interest
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Michelozzi, C., Di Leo, G., Galli, F. et al. Subependymal nodules and giant cell tumours in tuberous sclerosis complex patients: prevalence on MRI in relation to gene mutation. Childs Nerv Syst 29, 249–254 (2013). https://doi.org/10.1007/s00381-012-1892-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-012-1892-8