
Abstract
Objective
This article describes the clinical aspects for both operated and non-operated patients with a cloverleaf skull deformity treated in our service, focusing on hydrocephalus.
Methods
We describe 13 cases of cloverleaf skull deformity treated in our services between 1977 and 2008. Among them, ten were operated (9 out of 13 for the craniofacial stenosis and 7 out of 13 for hydrocephalus).
Results
Hydrocephalus was present in all patients with bilateral lambdoid stenosis. There was no case of hydrocephalus among the patients with unilateral or absent lambdoid stenosis. Associated malformations and severe faciostenosis were associated with higher mortality and morbidity.
Conclusion
The development of hydrocephalus seems to be closely related to a bilateral lambdoid stenosis. The optimal treatment must be tailored individually considering the degree of the malformation and the presence of complications and comorbidities.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The cloverleaf skull deformity (also known as Kleeblattschädel deformity and triphyllocephaly) is a very rare and very peculiar cranial malformation. The first publications describing this deformity appeared in the nineteenth century [1] but it was only in 1960 that Holtermüller and Wiedemann [2] published an article describing the Kleeblattschädel syndrome, assembling data from 11 previously published cases and one observation of their own. They originally described a deformity associating Kleeblattschädel deformity and high-grade hydrocephalus, extreme low position of the ears, face/skull deformities in the area of the orbits, nose and mandible, micromelia, pneumoencephalographic high-grade dyscrania, dysencephaly and lethal course. Since then many publications have shown that the cloverleaf skull malformation can be an isolated deformity but is often part of malformative syndromes related to fibroblast-growing factor receptor FGFR2 and FGFR3 anomalies (acrocephalosyndactyly—Crouzon, Pfeiffer type 2 and Apert syndromes, tanatophoric dysplasia and Beare–Stevenson cutis gyrata syndrome) or Boston type craniosynostosis, Carpenter syndrome, Cumming syndrome, osteoglophonic dysplasia, Saethre-Chotzen and chromosomal duplications of dup(4p), dup(13q), and dup(15q) amniotic bands [1, 3, 4].
The diagnosis of the cloverleaf deformity is purely phenotypical, based on the presence of a bitemporal (though sometimes asymmetrical) bulging. The typical aspect of this calvarial deformity is a severe stenosis of multiple sutures of the vault and the cranial base, resulting in a cloverleaf-shaped head with temporal bulging and inferiorly rotated ears. It is also often associated with hydrocephalus, hypoplastic midface and reduction of the cranial length [5–9]. The trilobed skull aspect is due to a bony constriction ring in the cranial base or at the level of the cranial sutural ring [10], leading to a cerebral frontal and temporal herniation. Exorbitism and some degree of prognathism are also common [8, 9]. Facial stenosis is frequently associated with this vault deformity and it is often the most dramatic aspect as a respiratory distress can be present even at birth. The orbital cavities can be very shallow leading to exposure keratopathy or even, in the most severe cases, to spontaneous enucleation.
Hydrocephalus and venous hypertension, along with craniocerebral disproportion and airway obstruction, are the important causes of intracranial hypertension in craniosynostotic children [11, 12]. The incidence of hydrocephalus in cloverleaf skull patients has always been considered elevated with as many as 100% in one series [13] and low as 20% in other [14].
This report describes the characteristics, evolutions and complications of 13 patients treated in our service that were diagnosed as having a cloverleaf skull. The identification of some clinical and radiological signs can help the neurosurgeon to determine the correct treatment and the surgical timing for this rare and challenging disease.
Patients and methods
We have retrospectively studied 13 affected patients treated in our service between 1977 and 2008. They correspond to 0.34% of the total of 3,773 patients diagnosed with craniosynostosis followed in our service during the same period. They all received the diagnosis of cloverleaf skull deformity based on the phenotypical aspects described above, identified on clinical and radiological exams (X-ray and CT scans in all cases, MRI in the most recent ones). They were nine males and four females with a mean age of presentation of 7.7 weeks (min 1.3 and max 44.6). Only two had prenatal diagnosis of craniostenosis. Five patients died during the follow-up. The mean follow-up of the eight other patients is 70 months. In our series, we have not considered the patients with isolated non-ossification of the temporal bone.
Four patients were identified as having Pfeiffer syndrome, three with Crouzon syndrome, one with auralcephalosyndactyly, one with an unknown syndrome and four with isolated cloverleaf malformation. Familial craniofacial malformation was only present in two cases (one patient with isolated cloverleaf skull deformity had one brother with Opitz C syndrome and another patient had a familial form of auralcephalosyndactyly syndrome).
The localization of the synostotic sutures was very variable (Table 1) with no relation with the degree of aesthetic malformation of the cranial shape or with the degree of facial stenosis. It has already been described that there is no relation between kleeblattschädel and a specific pattern of suture synostosis [3, 15]. The identification of an affected stenosis can be complicated, even during the surgery, when in presence of severe cranial lacunae, with the classic honeycomb aspect. All patients had some degree of reduction of the inter-ali sphenoidal angle, typical of the coronal stenosis. Most of the time, this reduction was very important causing a mephistopheles aspect with an elevated pterion.
All four patients considered as isolated cloverleaf deformity, the one diagnosed with Crouzon syndrome, had no malformation other than the skull deformity and had no familiar history of craniosynostosis. One patient with an unknown syndrome had an important dextroscoliosis associated with severe cardiopathy that ultimately led to the death of the child. Her chromosomal study showed no karyotypical anomaly. Four patients with Pfeiffer syndrome had mild to moderate limb deformities as typical of this syndrome. One had a severe bilateral genu varus. Two patients had the association of Crouzon syndrome and acanthosis nigricans, The patient with auralcephalosyndactyly had the characteristic familial and karyotypical signs of these diseases. All patients with Crouzon syndrome had a non-familial form and the diagnosis was based on the genetic studies.
Results
Craniofacial surgery
Major facial stenosis was present at birth in 5 of 13 patients, two of them requiring monobloc frontofacial advancement as a primary craniofacial surgery (one at 23 weeks of life and the other with only 6 weeks of life). As described in Table 2, two of them tolerated well this stenosis and the surgery was delayed. One last patient had a severe respiratory distress upon arrival (at the age of 9 weeks) and died few days later, before being stable enough to go to the operative room, despite all medical efforts. Shallow orbits were identified in nine cases. Two patients with severe exorbitism had recurrent spontaneous enucleation.
Ten children received surgical correction of the craniofacial deformity. Eight of them received a vault expansion surgery (fronto-parieto-occipital in seven cases and parieto-occipital in one) at a mean age of 28 weeks. Two patients with more severe facial stenosis were treated with frontofacial advancement at the age of 6 and 23 weeks. Five out of the eight patients that had a vault expansion as a first surgery were lately treated with frontofacial advancement. Two patients died prior to the surgery (one due to cardiac insufficiency and the other due to respiratory distress).
Hydrocephalus and posterior fossa malformation
The radiological techniques, as the surgical approaches, have evolved over the 30 years period of this study, and so the first three patients of this series did not receive a radiological examination precise enough to evaluate the posterior fossa in detail. Six out of the ten remaining patients had radiologic signs of Chiari malformation (three with Pfeiffer syndrome, one with Crouzon syndrome and two with isolated cloverleaf deformity).
All patients with documented Chiari malformation had bilateral lambdoid stenosis and they all had symptomatic ventricular dilatation (either at the moment of presentation or as a late manifestation). Nine out of 13 patients had ventricular dilatation, eight of them requiring ventricular drainage (Table 3). Six of the eight children that required ventricular evacuation had a Chiari malformation and one other patient had crowded posterior fossa (reduced fourth ventricle and basal cistern with bilateral lambdoid stenosis). One last patient (with an unknown syndrome and synostosis of the basal portion of both lambdoid sutures) had a hydrocephalus due to venous hypertension diagnosed with the identification of an equally elevated pressure in the ventricles and longitudinal sinus. In this case, the available radiological exams could not accurately identify a tonsilar herniation.
One patient with ventricular dilatation and no clinical signs of hydrocephalus had a sagittal and bicoronal synostosis with a first generation CT scan showing a reduced and crowded posterior fossa, but as in the previous case, a tonsilar herniation could not be radiologically evaluated with certitude. This patient was never treated for the ventricular dilatation.
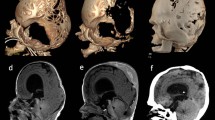
We have performed a posterior fossa decompression (consisting of occipital craniotomy with enlargement of the foramen magnum and the basal cistern) in three children at a mean age of 15.7 weeks, but in no case, there was a satisfactory reduction of the ventricular size or of the tonsilar herniation, even with a satisfactory radiological posterior fossa decompression (Fig. 1). An early posterior fossa decompression performed in two patients with Chiari syndrome failed to prevent hydrocephalus. One patient was treated with a third ventriculostomy that did not effectively control the hydrocephalus. In all cases, a ventricular drainage was the only efficient treatment for the hydrocephalus. Four patients with Chiari malformation received a ventricular drainage prior to or without posterior fossa decompression. In these patients, the tonsilar herniation was still present after the hydrocephalus treatment.

Left posterior fossa stenosis with Chiari malformation and hydrocephalus. Right postoperative imaging after occipital remodelling with the persistence of hydrocephalus
Only one out of four children without Chiari malformation developed hydrocephalus and had to be shunted (case 2). Even if there was no sign of tonsilar herniation in this hydrocephalic patient, his lambdoid sutures were synostotic and there was an apparent crowding of the posterior fossa with reduced fourth ventricle and basal cistern.
Among the four non-hydrocephalic patients, two had no lambdoid stenosis and two had unilateral lambdoid stenosis. One patient with unilateral lambdoid stenosis had a normal posterior fossa. In the other case of unilateral lambdoid stenosis, the posterior fossa could not be accurately evaluated due to the limitations of the exams available at the time of the hospitalization.
Long-term results
Cognitive evaluation was available for eight patients. Three non-hydrocephalic patients had a mean IQ of 94.3 (min 85, max 106), two of these children had major facial stenosis and one had a less severe form. Three hydrocephalic patients with mild facial stenosis had a mean IQ of 86 (min 74, max 95) and two other hydrocephalic children with severe facial stenosis were considered as severely retarded. Aesthetic long-term results were available in five patients, all submitted to total vault remodelling, and classified as previously described [16]. Three were considered as grade 1 (good), one as grade 2 (incomplete result) and one as grade 3 (poor result). There seemed to be no relation between the final aesthetic aspect and hydrocephalus or a syndromic condition.
Discussion
The cloverleaf skull malformation is a rare and heterogeneous deformity with different etiologies and degrees of severity [3, 10, 17]. Though many reports have been published since its description 50 years ago, the exact incidence of this deformity is difficult to establish, partially due to frequent interruptions of pregnancy (either spontaneous or medically induced). The diagnosis is based only on the phenotype, aided eventually by the tridimensional tomography, as the skull takes on a trilobed or cloverleaf appearance due to a bitemporal bulging in association with a frontal herniation through a constriction ring. Prenatal diagnosis has become possible using high-resolution ultrasonography [18] or MRI.
The Kleeblattschädel syndrome was classically considered to be the result of coronal and lambdoid intrauterine synostosis associated with congenital hydrocephalus. In our work, as in other publications, we have found that there is no specific pattern of sutural synostosis. We have also found that there is no relation between the synostotic suture and the degree of the aesthetic deformity [3, 9, 19, 20].
Hydrocephalus
Hydrocephalus is a condition that can be associated with cranyostenosis, especially in syndromic cases where it has been found in 12% to 15% of the cases [21, 22]. It may cause chronic intracranial hypertension with cognitive and visual deficits [23] and the association of suture stenosis and hydrocephalus may cause major intracranial hypertension. Premature fusion of the petro-occipital synchondrosis causes stenosis or atresia of the jugular foramen leading to venous hypertension and communicating hydrocephalus [11, 24]. Additionally, genetic abnormalities, as mutations on the FGFR1 and FGFR2 are suspected to be responsible for primarily narrowed sigmoid and jugular sinuses as a consequence of abnormalities in the endothelial proliferation [24]. Golabi [25] studied 250 cranosynostotic patients and found ten cases of hydrocephalus with five patients with kleeblattschädel deformity among them. Sadly, he failed to mention if there were kleeblattschädel deformity patients among the non-hydrocephalic ones. In our study, we have found nine children (69.2%) with ventricular dilatation, eight of them requiring treatment for hydrocephalus.
The overcrowding of the posterior fossa can deform the fourth ventricle leading to a non-communicating hydrocephalus [21, 26–29]. Cinalli et al [30] studied the relation between Chiari malformation and craniosynostosis and suggested that it was due to a precocious stenosis of the skull base sutures (sphenooccipital, petrous occipital and intraoccipital synchondrosis) causing a reduced posterior fossa and crowding the brain stem and the cerebellum in this narrow space. As those synchondroses are difficult to be evaluated surgically and radiologically, a synostosis of the lambdoid suture can be used as a reflection of the stenosis of the skull base sutures. This could be identified even on plain radiographies by the coup de serpe sign (Fig. 2), an acute angle of the posterior skull reflecting bilateral lambdoid stenosis. In our study, all nine patients with ventricular dilatation had bilateral lambdoid stenosis (Fig. 3) and none of the non-hydrocephalic ones had this bilateral synostosis. Two patients with unilateral lambdoid stenosis were not hydrocephalic and at least one did not have a reduced posterior fossa (Fig. 4).

Coup de serpe posterior fossa on the plain radiography. Chiari malformation on T1 sagittal MRI. Hydrocephalus

Left Bilateral lambdoid stenosis. (Center) Absence of hydrocephalus on prenatal MRI. (Right) hydrocephalus at age of 2 months

Unilateral lambdoid stenosis and absence of ventricular dilatation at age of 5 months
A posterior fossa decompression can efficiently liberate the foramen magnum and the basal cistern, correcting an eventual non-communicating hydrocephalus [30], but fails to treat jugular foramen stenosis and thus may be insufficient to treat the consequent hydrocephalus. A posterior fossa decompression was performed in three patients but failed to reduce the intracranial hypertension and the ventricular volume, suggesting that the venous hypertension is an important factor in the origin of the hydrocephalus in patients with cloverleaf skull deformity. Among the six patients with psychomotor evaluation, the four children with a better cognitive result were not hydrocephalic and the two with severe disabilities had ventricular dilatation treated with DVP shunts.
Surgical treatment
The surgical treatment of the cloverleaf deformity has evolved with time, from the excision of the synostotic sutures to a total vault reconstruction, facial advancement and posterior fossa decompression [1, 10, 15, 20, 26, 28, 31]. The surgical treatment aims to correct the vault deformity to reduce the intracranial pressure, to expand the shallow orbits to accommodate the globes, to open the nasopharyngeal airways to improve the breathing and to ameliorate the aesthetic deformity [32].
The ideal time for an elective craniosynostosis surgery is around the age of 6 months [31], for an ideal surgical weight and a fully developed hematologic system, but sometimes a child with cloverleaf skull deformity cannot wait for so long. In many cases, the surgical procedure has to be performed much sooner, imposed by a clinical condition. The midface hypoplasia often leads to upper airway stenosis and obstructive sleep apnea [10, 33]. In the case of severe airway obstruction, a tracheotomy might be realized in the first hours of life. The airway obstruction and shallow orbits can be corrected with frontofacial advancement and a symptomatic Chiari malformation can be treated with a posterior fossa decompression. Venes [34] successfully treated a cardiorespiratory distress in a patient with tonsilar herniation with a vault decompression. A ventricular drainage is often necessary, sometimes at birth. In this situation, a ventriculoperitoneal shunt seems to be the only efficient solution.
Most of the time, this complex craniosynostosis is corrected with a staged repair. Resnick [15] suggested an early and aggressive decompressive surgery, preferably at the first 48 h of life, followed or associated with frontofacial advancement. A precocious total vault reconstruction is also supported by the works of Zuccaro [14] and Goodrich [31]. More recently, Jarrahy [35] presented a series of 14 cloverleaf skull patients operated, finding an incidence of postoperative complications of 67% among patients treated with early cranial vault remodelling (mean age of 5.1 months, n = 3) and of 9% among those operated later (mean age of 13.2 months, n = 11). The aesthetic result was also more satisfactory among the patients operated later. It was then proposed as an optimal surgical treatment, a fronto-orbital advancement performed between 3 and 6 months of age and the posterior remodelling at the age of 1 year. Preuss et al [36] have described one child operated with craniectomy of the upper cranial vault at the age of 4 months followed by fronto-orbital advancement with strip cranioplasty at the age of 8 months with both aesthetical and cognitive good results. We usually perform a vault remodelling before 12 months of age (mean age of 8.5 months of age in this series) then later a frontofacial remodelling (mean age of 40 months).
Our study has shown that the prognosis and the surgical treatment are different for three groups of children with cloverleaf deformity (Table 4). First, there are some children that present the temporal bulging in the absence of major extracranial malformations, often associated with some degree of “frontal herniation” configuring the typical trilobed head shape of this syndrome. There can be no lambdoid stenosis and no hydrocephalus. The airway is not severely obstructed and these children have no significant respiratory distress. These patients have a better prognosis and the surgical treatment is not urgent, being focused on the prevention of intracranial hypertension, exorbitism or breathing impairment and the correction of the aesthetic deformity. The association with cranial base synchondrosis (that shall be suspected in the presence of bilateral lambdoid stenosis) may lead to reduced posterior fossa with tonsilar herniation, triventricular hydrocephalus and even a brain stem compression. The presence of increased inner pressures may aggravate the vault deformity [25]. In this situation, a ventriculoperitoneal shunt seems to be the only effective treatment for the hydrocephalus.
The second group comprises the children that present a more marked cloverleaf aspect with severe facial stenosis (important ocular exorbitism and respiratory distress due to the airway stenosis) and the primary treatment shall be focused on the airway liberation. Once again the presence of hydrocephalus and Chiari malformation must be suspected when in the presence of bilateral lambdoid stenosis. Most children with acrocephalosyndactyly and cloverleaf deformity are among these patients. The prognosis of these children is worst due to the respiratory distress and the frequent cranial hypertension that impose a more precocious (and often staged) surgical intervention.
Finally, there is a third group of children that presents major systemic malformation (often affecting long bones and the cardiorespiratory system) that are the most important, and often lethal, aspect of the disease. Some are so severely affected, as with the thanatophoric dwarfism, that no facial or vault surgery can be performed [7, 37].
Survival and prognosis
Five patients died during the period of the study. We had three precocious deaths (two secondary to major respiratory distress and one secondary to severe congenital cardiopathy). Cardiorespiratory distress is a frequent dangerous situation in these patients that often have the association of airway stenosis, intracranial hypertension and Chiari malformation. Two patients died due to massive surgical haemorrhage (during a monobloc frontofacial advancement).
Concerning long-term cognitive outcome, cloverleaf skull patients are generally considered to have a bad intellectual functioning outcome [33], but many exceptions have already been published. In 1972, Arseni et al [38] described a non-operated child that appeared cognitively normal for her age and Goh and Poon [39] later described a 37-year-old woman with cloverleaf skull deformity and chronic hydrocephalus that was the mother of two healthy children and had a normal married life. Lodge et al [10] suggested that the management of the respiratory insufficiency through the early and intermediate years is the determinant of the long-term outcome. Poor neurological development seems to be closely related not only to the chronic hypoventilation caused by the airway obstruction but also to the existence of intracranial hypertension. Long-term neuropsychological evaluation was available for eight of our patients. The three non-hydrocephalic children and the three hydrocephalic patients with mild facial stenosis had normal or near-normal results but the two hydrocephalic children with severe facial stenosis had a poor final cognitive outcome.
Conclusion
The cloverleaf deformity is not a uniform disease but a common feature of a great variety of diseases. The phenotypical aspect seems to have more importance than the pathogenetic one, as this malformation is the final result of a heterogenous group of diseases. Respiratory distress and systemic malformation are the most important features leading to a bad prognosis. Hydrocephalus in these patients seems to be closely related to the posterior fossa stenosis. Its association with bilateral lambdoid stenosis suggests that the constriction of the jugular foramens is an important factor in its origin. Aggressive and precocious treatment of the hydrocephalus and the cranial hypertension in selected patients may lead to good long-term results.
References
Cohen MM Jr (2005) Editorial: perspectives on craniosynostosis. Am J Med Genet A 136A:313–326
Holtermueller K, Wiedemann HR (1960) The clover-leaf skull syndrome. Med Monatsschr 14:439–446
Cohen MM, MacLean RE (2000) Craniosynostosis: diagnosis, evaluation, and management. Oxford University Press, New York
Robin NH, Falk MJ, Haldeman-Englert CR (1993) FGFR-related craniosynostosis syndromes. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds) GeneReviews. University of Washington, Seattle
Bonucci E, Nardi F (1972) The cloverleaf skull syndrome. Histological, histochemical and ultrastructural findings. Virchows Arch A Pathol Pathol Anat 357:199–212
O’Broin ES, O’Keefe M, Allcutt D, Earley MJ (1997) Cloverleaf skull anomaly with extreme orbitostenosis. J Craniofac Surg 8:75–77
Oyamada MK, Ferreira HS, Hoff M (2003) Pfeiffer syndrome type 2—case report. Sao Paulo Med J 121:176–179
Goodrich JT (2005) Skull base growth in craniosynostosis. Childs Nerv Syst 21:871–879
Witt PD, Hardesty RA, Zuppan C, Rouse G, Hasso AN, Boyne P (1992) Fetal kleeblattschadel cranium: morphologic, radiographic, and histologic analysis. Cleft Palate Craniofac J 29:363–368
Lodge ML, Moore MH, Hanieh A, Trott JA, David DJ (1993) The cloverleaf skull anomaly: managing extreme cranio-orbitofaciostenosis. Plast Reconstr Surg 91:1–9, discussion 10–14
Hayward R (2005) Venous hypertension and craniosynostosis. Childs Nerv Syst 21:880–888
Taylor WJ, Hayward RD, Lasjaunias P, Britto JA, Thompson DN, Jones BM, Evans RD (2001) Enigma of raised intracranial pressure in patients with complex craniosynostosis: the role of abnormal intracranial venous drainage. J Neurosurg 94:377–385
Partington MW, Gonzales-Crussi F, Khakee SG, Wollin DG (1971) Cloverleaf skull and thanatophoric dwarfism. Report of four cases, two in the same sibship. Arch Dis Child 46:656–664
Zuccaro G, Dogliotti P, Bennum R, Monges J (1996) Treatment of cloverleaf skull syndrome. Childs Nerv Syst 12:695–698
Resnick DK, Pollack IF, Albright AL (1995) Surgical management of the cloverleaf skull deformity. Pediatr Neurosurg 22:29–37, discussion 238
Renier D, El-Ghouzzi V, Bonaventure J, Le Merrer M, Lajeunie E (2000) Fibroblast growth factor receptor 3 mutation in nonsyndromic coronal synostosis: clinical spectrum, prevalence, and surgical outcome. J Neurosurg 92:631–636
Esmer MC, Rodriguez-Soto G, Carrasco-Daza D, Iracheta ML, Del Castillo V (2000) Cloverleaf skull and multiple congenital anomalies in a girl exposed to cocaine in utero: case report and review of the literature. Childs Nerv Syst 16:176–179, discussion 180
Kalache KD, Lehmann K, Chaoui R, Kivelitz DE, Mundlos S, Bollmann R (2002) Prenatal diagnosis of partial agenesis of the corpus callosum in a fetus with thanatophoric dysplasia type 2. Prenat Diagn 22:404–407
Hayward R, Jones B, Dunaway D (2004) The clinical management of craniosynostosis. MacKeith, London
Gosain AK, Moore FO, Hemmy DC (1997) The kleeblattschadel anomaly in Apert syndrome: intracranial anatomy, surgical correction, and subsequent cranial vault development. Plast Reconstr Surg 100:1796–1802
Cinalli G, Sainte-Rose C, Kollar EM, Zerah M, Brunelle F, Chumas P, Arnaud E, Marchac D, Pierre-Kahn A, Renier D (1998) Hydrocephalus and craniosynostosis. J Neurosurg 88:209–214
Di Rocco F, Juca CE, Arnaud E, Renier D, Sainte-Rose C (2010) The role of endoscopic third ventriculostomy in the treatment of hydrocephalus associated with faciocraniosynostosis. J Neurosurg Pediatr 6:17–22
Marchac D, Renier D, Flandin-Bléty C (1982) Chirurgie cranio-faciale des craniosténoses. Medsi
Sainte-Rose C, LaCombe J, Pierre-Kahn A, Renier D, Hirsch JF (1984) Intracranial venous sinus hypertension: cause or consequence of hydrocephalus in infants? J Neurosurg 60:727–736
Golabi M, Edwards MS, Ousterhout DK (1987) Craniosynostosis and hydrocephalus. Neurosurgery 21:63–67
Angle CR, McIntire MS, Moore RC (1967) Cloverleaf skull: Kleeblattschadel-deformity syndrome. Am J Dis Child 114:198–202
Raybaud C, Di Rocco C (2007) Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: a review. Childs Nerv Syst 23:1379–1388
Thompson DN, Hayward RD, Harkness WJ, Bingham RM, Jones BM (1995) Lessons from a case of kleeblattschadel. Case report. J Neurosurg 82:1071–1074
Shiroyama Y, Ito H, Yamashita T, Nakano S, Kurokawa Y (1991) The relationship of cloverleaf skull syndrome to hydrocephalus. Childs Nerv Syst 7:382–385
Cinalli G, Chumas P, Arnaud E, Sainte-Rose C, Renier D (1998) Occipital remodeling and suboccipital decompression in severe craniosynostosis associated with tonsillar herniation. Neurosurgery 42:66–71, discussion 71–63
Goodrich JT, Staffenberg DA (2004) Plastic techniques in neurosurgery. Thieme, New York
Kroczek RA, Muhlbauer W, Zimmermann I (1986) Cloverleaf skull associated with Pfeiffer syndrome: pathology and management. Eur J Pediatr 145:442–445
Moore MH, Cantrell SB, Trott JA, David DJ (1995) Pfeiffer syndrome: a clinical review. Cleft Palate Craniofac J 32:62–70
Venes JL (1988) Arnold-Chiari malformation in an infant with Kleeblattschadel: an acquired malformation? Neurosurgery 23:360–362
Jarrahy R, Kawamoto HK, Keagle J, Dickinson BP, Katchikian HV, Bradley JP (2009) Three tenets for staged correction of Kleeblattschadel or cloverleaf skull deformity. Plast Reconstr Surg 123:310–318
Preuss M, Stein M, Neubauer BA, Schaaf H, Howaldt HP, Nestler U, Christophis P (2010) About the operative management and post-operative neural development of patients with cloverleaf skull deformity. Childs Nerv Syst 26:1211–1218
Kozlowski K, Jequier S, Sillence D, Moir DH (1987) Cloverleaf skull and bone dysplasias (report of four cases). Australas Radiol 31:309–314
Arseni C, Horvath L, Ciurea V (1972) Cloverleaf skull. Acta Neurochir (Wien) 27:223–230
Goh KY, Poon WS (1997) A case of adult kleeblattschadel. Acta Neurochir (Wien) 139:1088–1089
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Machado, G., Di Rocco, F., Sainte-Rose, C. et al. Cloverleaf skull deformity and hydrocephalus. Childs Nerv Syst 27, 1683–1691 (2011). https://doi.org/10.1007/s00381-011-1508-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-011-1508-8