
Abstract
Introduction
The mechanisms responsible for cognitive decline after traumatic brain injury (TBI) in pediatric patients are poorly understood. The present study examined the potential role of synaptic alterations in this process by using an animal model of immature head injury to define the impact of TBI on expression of the synaptic protein, synaptophysin.
Materials and methods
After craniotomy, TBI was induced in postnatal day 17 (PND17) rats using controlled cortical impact delivered to the left hemisphere. NeuN, a neuronal marker, and synaptophysin expression were examined 1 day, 1 week, and 1 month after injury by immunohistochemistry and immunoblotting.
Results
There were significant decreases in both NeuN and synaptophysin after 1 day and 1 week but not 1 month after injury within the hippocampus and neocortex adjacent to the impact site compared to sham-injured controls. The decrease in synaptophysin and NeuN was also noted in the contralateral hippocampus by 1 day after injury and in the contralateral neocortex by 1 week, indicating that changes in protein expression were not solely localized to the injury site but occurred in more distant regions as well.
Discussion
In conclusion, the decrease and recovery in synaptophysin parallel the cognitive changes that occur after experimental TBI in the PND17 rat, which suggests that changes in this protein may contribute to cognitive declines after injury. The results also suggest that, in spite of the focal nature of the impact, diffuse alterations in protein expression can occur after immature TBI and may contribute to the subsequent cognitive dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) remains a serious pediatric health problem with a reported incidence in children of 230/100,000 [20]. Approximately 10–15% of those cases are severe, resulting in death or permanent brain damage in 10,000–30,000 children per year [7, 22]. Even more children suffer from only mild or moderate brain injury but remain neurocognitively disabled [5, 21]. Considering the large number of children seriously injured each year, enhanced treatment methods targeted at neurocognitive recovery could not only improve quality of life of the affected individuals but also improve their productivity and reduce the economic impact on society as a whole. However, devising new therapeutic strategies to improve outcome requires a thorough understanding of the impact of the primary brain injury and the secondary events that lead to long-term dysfunction. While there have been a great number of studies on the effects of TBI on the adult brain, there have been relatively few studies on the developing brain, so current understanding of the impact of TBI on the immature brain is limited.
The response of the immature brain to injury is likely different than that of the adult because many developmental processes are subject to disruption by insults such as TBI. A number of processes, including generation of new neurons, myelination, and synaptogenesis, continue postnatally. Postnatal neurogenesis occurs within the cerebellum and dentate gyrus (DG) of the hippocampus in both the human and rodent brain [3, 4, 32]. Gliogenesis and myelination start prenatally and continue through adolescence in both humans and rodents [4, 15, 17, 32]. Generation and maturation of synapses continues for at least 21 days in rats and through puberty in human and nonhuman primates [6, 18, 26, 31, 39]. Because the temporal sequence of ontogenic processes is critical for production of effective neural circuity, disruption of developmental processes by TBI at certain times during development could have particularly devastating, long-lasting consequences. The vulnerability of developmental processes to disruption by TBI may account for the finding that children under 4 have more severe cognitive dysfunction after brain trauma than older children and adults [13, 23, 27]. Similarly, although excitatory amino acid receptor blockers often improve outcome after TBI in adult animals, these agents may exacerbate damage in the more immature brain after ischemic injury and TBI [1, 16, 30, 37].
For the present study, we hypothesized that TBI would have a negative impact on neuronal connections in the immature brain by adversely affecting synaptogenesis and synaptic maturation. The study sought to determine the effect of TBI on expression of synaptophysin (SYN), a presynaptic protein, in the immature, postnatal day 17 (PND17) rat brain. At this age, the rat brain is roughly comparable to that of a young child based on indices such as brain growth, cellular proliferation, electroencephalography patterns, and synaptogenesis [14]. Furthermore, as noted above, synaptogenesis and synaptic maturation are still occurring at this time.
Materials and methods
Animals
All experiments were done using PND17 male and female rat pups. The pups were housed with their mothers and had free access to food and water before and after injury. They were weaned on PND21. For the immunohistochemical studies, a total of three injured, three naïve, and one sham-treated animals were used for each time point. There were no clear differences between the naïve and sham-treated animals, so they were combined into a single control group to increase statistical power. A total of four injured and four sham-treated animals were used for each time point of the immunoblot analyses.
Injury
The animals were anesthetized with 1–2% isoflurane and placed in a stereotactic head frame. After a midline incision, a 6-mm-wide craniotomy was made in the skull with a dental drill. A controlled cortical impact (CCI) device was used to generate a mechanical injury between the bregma and lambda (6-mm tip, 4 m/s, deflection = 2 mm). Animals assigned to the sham group underwent the same surgery but without the injury. Those in the naïve group were not manipulated. Animals were observed daily for behavioral and neurologic alterations.
Tissue preparation
At 1 day, 1 week, and 1 month after injury, the rats were anesthetized with pentobarbital and killed by intracardiac perfusion with phosphate-buffered saline (PBS) followed by 10% buffered formalin. The brains were then removed, postfixed in formalin overnight, and transferred to 30% sucrose. Once the brains were infiltrated with sucrose, they were sliced sagitally on a sliding microtome, and 40-μm-thick sections were collected every 240 μm throughout the width of the brain. The slices were placed in a glycol-based cryoprotectant solution consisting of 30% sucrose, 30% ethylene glycol, 1% polyvinyl-pyrollidone in a 0.05-M sodium phosphate buffer (pH 7.2), and stored at −20°C until undergoing immunohistochemical staining.
Immunohistochemistry
Tissue sections were stained for the presence of both SYN and NeuN. All antibodies and detection reagents were diluted in PBS except where specified otherwise. Free-floating sections were rinsed in PBS and then postfixed for 10 min in 10% formalin. After an additional PBS rinse, they were placed in 2% goat serum with 0.2% Triton-X for 1 h at room temperature to block nonspecific binding. The sections were then rinsed in PBS followed by overnight incubation in a solution containing a 1:200 dilution of the SYN antibody (Santa Cruz Biotech, Santa Cruz, CA). Binding of the SYN antibody was then detected using a biotinylated anti-rabbit antibody (Vector Labs, Burlingame, CA; 1:100 dilution) and Alexa 594-conjugated strepavidin (Invitrogen, Carlsbad, CA; 1:100). The fluorescent signal was amplified using biotinylated anti-strepavidin (Vector Labs; 1:100) followed by an additional incubation in Alexa 594-conjugated strepavidin (1:100).
The same sections were subsequently stained for expression of NeuN. After overnight incubation at 4°C in a solution containing a 1:500 dilution of antibody to NeuN (Chemicon, Temecula, CA), the sections were rinsed in PBS and incubated overnight in a solution containing a 1:1,000 of dilution Alexa 488-conjuated goat anti-mouse antibody (Invitrogen). The tissues were then mounted on slides with a gelatin subbing solution, dehydrated in an ascending alcohol series, and mounted in a Tris-buffered glycerol solution containing 1% p-phyenylenediamine to reduce fluorescent quenching.
Image analysis
SYN and NeuN expression were evaluated qualitatively and semiquantitatively. For semiquantitative analysis, microscopic images of the hippocampus directly ventral to the trauma site, the neocortex directly adjacent to the trauma site, and corresponding regions of the contralateral hemisphere were collected under epifluorescence using filters optimized for the Alexa fluorophores used in these experiments (Fig. 1). To determine background staining of regions without appreciable NeuN or SYN expression, images of white matter region of the internal capsule were also collected. All images were collected using a 10× objective by a CCD camera (Axiocam, Carl Zeiss Microimaging, Thornwood, NY) and transferred to a computer workstation. The images were then processed and analyzed using an automated macro created with the KS300 image analysis software package (Zeiss).

Schematic diagram (a) and microphotographs (b) showing the regions of interest used in evaluating the expression of synaptophysin and NeuN based on immunostaining. As illustrated in the schematic, the controlled cortical impact device was centered dorsal to the hippocampus (Hp). Locations of the neocortex (Nc), corpus callosum (CC), caudate (Cd), lateral ventricle (LV), and cerebellum (Cb) are also indicated. The white and black outlines (a) delineate the regions shown in the photographs to the right (b). Within the neocortex, three layers could be readily distinguished based on cell density. Superficial and deep layers with relatively high cell density were separated by a lower density, intermediate layer. Rectangular ROIs were drawn in each of these three regions. In the hippocampus, ROIs were drawn to outline sections of CA1 and CA3 of Ammon’s horn and also the dentate gyrus (DG). The hippocampal ROIs surrounded and extended approximately 50 μm beyond the pyramidal and granular cell layers
To correct for variations in intensity within the image because of alterations in exposure time or inhomogeneity of the fluorescent light source, each image was normalized on a pixel-by-pixel basis to the exposure time and to a baseline image, which was an image collected under epifluorescent illumination with only a blank slide in the microscope. The green channel of the green fluorescent images, which showed NeuN staining, was then combined with the red channel of the red fluorescent images, which showed SYN antibody staining, into a single color image. For analysis of staining within the hippocampus, rectangular regions of interest (ROIs) were drawn around the pyramidal layers of CA1 and CA3 and the granular layer of the dorsal blade of the DG (Fig. 1b). The ROIs were drawn so that they also encompassed approximately 50 μm both dorsal and ventral to the pyramidal and granular layers. Each image was then automatically segmented into neuronal (high levels of green fluorescence) and nonneuronal regions, and the thickness of the pyramidal and granular layers were calculated based upon this segmentation. In addition, average green fluorescent intensity was measured within the neuronal region, and average red fluorescent intensity was measured within the nonneuronal region as measures of NeuN and SYN staining, respectively. For analysis of background staining in regions with little or no NeuN and SYN expression, a rectangular ROI was drawn over the internal capsule and average green and red intensity measured. Intensity of the fluorescence in the hippocampal regions was normalized to the intensity of the background region to correct for nonspecific staining and potential decreases in overall fluorescent intensity during storage. For analysis of antigen expression in the cortex after injury, the neocortex was divided into three layers of equal thickness, superficial, intermediate, and deep, which covered the region between the brain surface and the subcortical white matter (Fig. 1b). ROIs were drawn in each of the three layers, and the average green and red intensity in the ROIs were calculated and normalized to the intensity of the background ROIs.
Immunoblotting
Immediately after euthanasia, tissue samples were collected from the neocortex directly adjacent to the trauma site and from the entire hippocampus. Samples were also collected from comparable regions of the contralateral hemisphere. The samples were immediately frozen in liquid nitrogen and stored at −80°C for later processing.
For protein extractions, tissue samples were homogenized in a buffer (pH 7.5) containing 167 mM NaCl, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, and a protease inhibitor cocktail (Calbiochem/EMD Biosciences, San Diego, CA). Protein concentration was determined using BCA Protein Assay Kit (Pierce, Rockford IL).
For each gel, protein samples from both the ipsilateral and contralateral side of one region from an injured animal were paired with similar samples from an uninjured, control animal. Samples were loaded onto a 4–20% Tris-Glycine gel (Invitrogen, Carlsbad, CA) and electrophoretically separated at 190 V for 1 h. Proteins were then transferred to a PVDF membrane using a semidry system run at 150 mA for 1.5 h. The membrane was then incubated with 10% milk in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h to block nonspecific binding and then incubated with anti-SYN antibody (Chemicon) diluted 1:1,000 in TBST with 5% milk (TBST-M) for 1 h at room temperature. The membrane was then washed in TBST-M and subsequently incubated in a solution containing HRP-conjugated rabbit anti-mouse IgG (Jackson ImmunoResearch Laboratory, West Grove, PA), diluted 1:5,000 in TBST-M for 1 h at room temperature. After rinsing in TBST, binding of the antibodies to the membrane was detected using the ECL Western Blotting Detection Kit (Amersham Biosciences, Buckinghamshire, England) according to the directions of the manufacturer.
X-ray film-based images of the immunoblots were scanned and transferred to a computer workstation for densitometric analysis. Density and size of the bands of each blot were measured using the ImageJ image analysis software program available from the National Institutes of Health (NIH). The relative amount of protein at a given pixel within the image was based on the (A) absorbance of light from the scanner using the formula, A = Log (P/P 0), where P represents the transmitted light corresponding to a given pixel, and P 0 represents the incident light, which was measured based on the average pixel intensity for a blank region of the gel that contained no apparent protein and that was adjacent to the protein band being analyzed. This formula was based on Beer’s Law, which states that the absorption of light by a translucent material is proportional to the concentration of a given compound within it. The value of A was summed over all the pixels for a particular protein band to estimate the total amount of SYN or NeuN within a given sample.
There can be significant blot-to-blot differences because of a variety of factors, such as alterations in electrophoretic conditions, changes in antibody binding, and variations in exposure, so only samples run on the same gel were directly compared, and the ratios of the NeuN or SYN for the injured animal to that of the control animal sample were calculated for the ipsilateral and contralateral hemisphere on each gel, and these values were used for further statistical analyses.
Statistical analysis
The impact of CCI on NeuN and SYN expression as a function of time after injury as measured by immunohistochemistry was tested for statistical significance by analysis of variance (ANOVA) using the JMP statistical software program (SAS Institute, Cary, NC). The factors included in the linear model used in the ANOVA were treatment, time after injury, and the interaction of these two terms. If any of the factors proved to be statistically significant (p < 0.05), a test of contrasts was used to compare differences in treatment groups at each individual time point. Statistical comparisons were carried out separately for the injured and contralateral hemispheres. In the case of immunoblotting, a 95% confidence interval for the ratio of protein levels in the CCI animals relative to that in controls was established. This value was then compared to one, which would be the value if the two groups were equivalent.
Results
At 1 day after injury, there was significant disruption of the neocortex in the injured hemisphere, whereas the underlying hippocampus was relatively intact (Fig. 2). By 1 week after injury, there was significant retraction of the neocortex, and a full thickness defect was present at the injury site. In addition, the hippocampus directly ventral to the impact was reduced in size or absent altogether. No gross histologic changes were evident in the sham-treated animals. In spite of the clear histologic changes in the injured animals, behavioral changes or neurologic deficits were rare with less than 5% of all animals showing a mild head tremor that resolved by 1–2 days after injury.

Expression of SYN (red stain) and NeuN (green stain) in the hippocampus of the injured hemisphere of both controls (a, c, e) and CCI-treated (b, d, f) animals at 1 day (a, b), 1 week (c, d), and 1 month (e, f) after injury. In the CCI-treated animals, there were occasional losses of NeuN staining in the pyramidal and granular layers of Ammon’s horn and the DG (b). By 1 week after injury, the hippocampus directly below the injury site was absent, and there were no obvious alterations in SYN and NeuN expression in the remaining hippocampal regions
With regard to protein expression after injury, there were significant decreases in both SYN and NeuN 1 day after CCI within both the hippocampus and cortex of the injured hemisphere (Fig. 3). Most of these decreases resolved over time. By 1 month after injury, levels of NeuN within the hippocampus and of both NeuN and SYN within the cortex of injured animals were not significantly different from levels in control animals. In contrast, SYN levels remained decreased within the hippocampus at 1 month. Unexpectedly, there were also significant decreases of NeuN and SYN within the hemisphere contralateral to the injury. The decreases were acute within the hippocampus, occurring 1 day after injury, but they were delayed within the cortex, not appearing until 1 week after injury. Like the alterations in NeuN and SYN within the injured hemisphere, the changes in the contralateral hemisphere generally resolved by 1 month after injury.

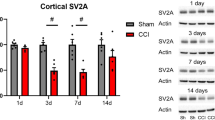
Analysis of expression of SYN and NeuN by immunoblot following CCI. Representative immunoblots demonstrating expression of SYN (left) and NeuN (right) in sham-treated controls to that in CCI-treated animals at day 1 after injury are shown at the top (a). Expression was measured in the hippocampus (top blots; a) and cortex (bottom blots; a) within both the injured (INJ) and the contralateral hemispheres (CONTRA). Quantitative densitometric analysis of the immunoblots demonstrated that there was a reduced expression of both SYN and NeuN within the cortex of the injured hemisphere and within the hippocampus of both the injured and the contralateral hemisphere 1 day after injury when compared to sham-treated controls (b). The expression generally increased over time so that the differences were minimal by 1 month after injury. For each gel, samples from a sham-treated animal were paired with a CCI-treated animal, and the ratio of expression in the injured to the uninjured animal was calculated. The dotted line indicates the line of unity, which is the expected value if the expression in the two animals is equivalent. Data points that differ significantly from the unity are marked by an asterisk (p < 0.05, n = four pairs of animals per data point)
Despite the alterations in NeuN and SYN detectable by Western blotting, there were no significant treatment-related changes in expression of these two antigens by immunohistochemistry within the hippocampus and neocortex of either hemisphere (data not shown). Nevertheless, there were developmental increases in SYN expression within the CA1, CA3, and DG regions of the hippocampus of control animals that either reached or approached statistical significance (p < 0.05, p = 0.054, and p = 0.05, respectively; Fig. 4). In addition, immunohistochemical analyses demonstrated that CCI caused a significant decrease in the width of the pyramidal layer of CA1 and CA3 and the granular layer of DG 1 day and 1 week after injury (Fig. 5). As mentioned earlier, by 1 month after injury, the region of the hippocampus directly ventral to the lesion was commonly absent. In the remaining hippocampus, there were no significant differences at that time in the width of the neuronal layers.

Expression of SYN within CA1, CA3, and the DG of the hippocampus of control animals increased as a function of age from postnatal day 8 to postnatal day 40. Brain sections were immunohistochemically stained for the expression of SYN, and the sections were then subjected to a computer-based analysis of staining intensity. Each point represents the average of four animals

Width of the pyramidal layers of CA1 (top) and CA3 (middle) and the granular layer of the DG (bottom) as a function of time after injury. These hippocampal layers were significantly reduced in thickness 1 day after CCI within the injured but not the CONTRA hemisphere when compared to controls (p < 0.05, ANOVA followed by a test of contrasts). Each point represents the average of three to four animals
Discussion
In an effort to design better therapeutic interventions and to optimize current ones, we sought to further our understanding of the pathologic changes that occur in children after TBI. Toward that end, we have been using an experimental rat model to delineate the impact of TBI on synaptogenesis and synaptic connectivity in the immature brain. Our current results demonstrate that experimental TBI reduces synaptic protein expression in the acute (1 day after injury) and subacute (1 week after injury) periods. While these results alone do not directly lead to potential therapeutic interventions, they do provide a molecular marker that can be monitored to evaluate the impact of new therapies on the development of damage. In addition, the results show one trauma-induced change, a decreased expression of a synaptic protein, that could contribute to the acute cognitive dysfunction and recovery 1 month later after TBI [36].
The current study employed two complementary approaches, optical densitometry of immunohistochemical sections and immunoblotting, to assess SYN and NeuN levels after injury. Immunoblotting allows for the direct quantitative comparison of protein levels of two or more samples. While immunohistochemical analyses may be less accurate than immunoblotting in determining the magnitude of changes in protein expression, immunohistochemistry has greater resolution when trying to detect region-specific alterations. However, there are also differences between the two techniques that could lead to disparate results. In the present study, there were decreases in SYN and NeuN expression based on immunoblotting that were not detected by immunohistochemistry, and several factors may contribute to this apparent disparity. For one, immunoblotting generally detects only intact proteins of the correct molecular weight, whereas immunohistochemistry may detect degraded or modified proteins as long as the epitopes that are recognized by the antibodies are present and in the proper conformation. Although we took great care to allow for variations in background staining and decreases in fluorescence that may occur over time, the sensitivity of optical densitometry to changes in SYN expression is reported to be limited relative to other methods [9, 10]. Nevertheless, we did detect an increase in SYN over time within the hippocampus of control animals, and the time course was consistent with synaptogenesis continuing up until PND21 in rats [26]. This finding suggests that reduced sensitivity of the immunohistochemical method alone was probably not responsible for the different findings of the two methods, and it emphasizes the importance of using caution when interpreting data provided by only one methodological approach.
The present study focused on synaptic alterations after TBI in the immature brain using SYN as a marker. One reason for focusing on this aspect of development is that synaptogenesis continues well into the postnatal period, and interference with this process could have devastating and long-lasting consequences on the establishment of effective neural circuits. SYN is a 38-kDa integral membrane protein found in presynaptic vesicles [8, 19, 41]. The expression pattern of SYN appears to parallel the development of new synapses during development [40]. Based on such results, a number of studies have used the measurement of SYN to assess synaptogenesis after various insults such as fluid percussion, deafferentation, and hypoperfusion [25, 28, 33]. Our study included assessment of NeuN, which is commonly used as a neuron-specific antigen. The initial reason for measuring this protein was to provide a way to differentiate alterations in synaptic protein levels at the cellular level from changes due only to neuron loss. Indeed, there were changes in not only SYN but also NeuN expression after injury, so some of the decreases in SYN may be related to cell loss. However, the changes in NeuN expression occurred not only on the side of impact, where there were clear losses of neuronal populations as shown by decreased thickness of the pyramidal and granular layers of the hippocampus (Fig. 5), but also on the contralateral side where there were no other obvious changes in neuron number. These results indicate that cellular expression of NeuN itself can change in response to brain injury regardless of changes in cell number, a finding that has been reported by others after neuronal damage because of ischemia and axotomy [29, 38]. NeuN is a poorly characterized protein, so the functional significance of this change is not clear, but it may reflect changes in phosphorylation status, which can affect antibody binding [24]. As both NeuN and SYN decreased in parallel, the alterations in SYN could represent either loss of specific proteins or a diffuse decrease in protein synthesis after injury. Our own preliminary data (not shown) examining changes in other proteins such as actin in PND7 animals after injury suggest that NeuN and SYN can be altered while other cellular proteins remain unchanged.
In addition to bilateral changes in NeuN after injury, there were also bilateral changes in SYN, so that altered protein expression was affected in regions distant from those directly surrounding the point of impact. These bilateral alterations took place in both the neocortex and hippocampal formation. The hippocampus is a brain region that is well-recognized for its critical role in learning and memory, so the present findings are consistent with the cognitive deficits that we have measured in young animals after TBI by Morris water maze testing [36]. While selective learning and memory deficits can occur when only one side of the hippocampal formation is lost or removed, such as is done in the treatment of refractory temporal lobe epilepsy, more severe deficits such as global amnesia occur after bilateral damage [12, 34]. Considering this, the fact that the changes in SYN occurred bilaterally supports the potential role of synaptic alterations in cognitive deficits. Our data showed that SYN expression recovers by 1 month after injury, which is also consistent with our own results that PND17 animals recover from TBI-induced cognitive deficits by 1 month [36]. The present findings are further supported by a number of reports using mature animals that have demonstrated an association between decreased cognitive performance and reduced SYN levels [10, 11, 25, 35]. There are several potential mechanisms by which changes in SYN could be conveyed to the contralateral hemisphere. One possibility is that neural circuits traveling through commissural connections between the ipsilateral and contralateral hippocampus transmit signals of the damage to the opposite hemisphere. Thus, the changes may be similar to diaschisis, in which alterations in blood flow and metabolism after a focal stroke are transmitted to distant regions, apparently via neuronal pathways [2]. Another possibility is that soluble factors released from the injured hippocampus may diffuse to the other side. Finally, shifts in the hemisphere opposite to the side of impact and compression against the skull (“contrecoup”) could contribute to contralateral changes. In any case, our results indicate that, although experimental TBI may be focal, the induced alterations can be widespread in the immature brain, and evaluation of regions distal to the site of injury may be required to fully understand the pathogenesis of damage in the immature brain.
References
Adelson PD, Dixon CE, Davis DS, Rodriguez, AG, Tran MP, Jenkins LW, Kochanek PM (2001) Differential age-at-injury effect of NMDA blockade on outcome following controlled cortical impact in immature rats. J Neurotrauma 18:1143
Andrews RJ (1991) Transhemispheric diaschisis. A review and comment. Stroke 22:943–949
Bayer SA (1980) Development of the hippocampal region in the rat. II. Morphogenesis during embryonic and early postnatal life. J Comp Neurol 190:115–134
Bayer, SA, Altman J, Russo RJ, Zhang X (1993) Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology 14:83–144
Beers SR (1992) Cognitive effects of mild head injury in children and adolescents. Neuropsychol Rev 3:281–320
Bourgeois JP, Goldman-Rakic PS, Rakic P (1994) Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex 4:78–96
Bruce DA, Schut L, Bruno LA, Wood JH, Sutton LN (1978) Outcome following severe head injuries in children. J Neurosurg 48:679–688
Buckley K, Kelly RB (1985) Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol 100:1284–1294
Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR (1996) Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol 25:821–828
Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M (1998) Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging 19:599–606
Chen KS, Masliah E, Mallory M, Gage FH (1995) Synaptic loss in cognitively impaired aged rats is ameliorated by chronic human nerve growth factor infusion. Neuroscience 68:19–27
Deweer B, Pillon B, Pochon JB, Dubois B (2001) Is the HM story only a ‘remote memory’? Some facts about hippocampus and memory in humans. Behav Brain Res 127:209–224
Fletcher JM, Ewing-Cobbs L, Francis DJ, Levin HS (1995) Variability in outcomes after traumatic brain injury in children: a developmental perspective. In: Broman SH, Michel ME (eds) Traumatic head injury in children. Oxford University Press, New York, pp 3–21
Hagberg H, Bona E, Gilland E, Puka-Sundvall M (1997) Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatr Suppl 422:85–88
Hunter SF, Leavitt JA, Rodriguez M (1997) Direct observation of myelination in vivo in the mature human central nervous system. A model for the behaviour of oligodendrocyte progenitors and their progeny. Brain 120(Pt 11):2071–2082
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74
Iwasaki N, Hamano K, Okada Y, Horigome Y, Nakayama J, Takeya T, Takita H, Nose T (1997) Volumetric quantification of brain development using MRI. Neuroradiology 39:841–846
Jacobson M (1991) Formation of dendrites and development of synaptic connections. In: Jacobson M (ed) Developmental neurobiology. Plenum, New York, pp 223–284
Jahn R, Schiebler W, Ouimet C, Greengard P (1985) A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proc Natl Acad Sci USA 82:4137–4141
Kraus JF, Fife D, Conroy C (1987) Pediatric brain injuries: the nature, clinical course, and early outcomes in a defined United States’ population. Pediatrics 79:501–507
Levin HS (1985) General considerations and neurobehavioral recovery. Part II. Neurobehavioral recovery. In: Becker DP, Povlishock JT (eds) Central nervous suystem status report. NINCDS and NIH, Washington, D.C., pp 281–299
Levin HS, Ewing-Cobbs, Eisenberg HM (1995) Neurobehavioral outcome of pediatric closed head injury. In: Broman SH, Michel ME (eds) Traumatic head injury in children. Oxford University Press, New York, pp 70–94
Levin HS, Aldrich EF, Saydjari C, Eisenberg HM, Foulkes MA, Bellefleur M, Luerssen TG, Jane JA, Marmarou A, Marshall LF, Al E (1992) Severe head injury in children: experience of the traumatic coma data bank. Neurosurgery 31:435–443, discussion 443–434
Lind D, Franken S, Kappler J, Jankowski J, Schilling K (2005) Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J Neurosci Res 79:295–302
Liu HX, Zhang JJ, Zheng P, Zhang Y (2005) Altered expression of MAP-2, GAP-43, and synaptophysin in the hippocampus of rats with chronic cerebral hypoperfusion correlates with cognitive impairment. Brain Res Mol Brain Res 139:169–177
Lohmann SM, Ueda T, Greengard P (1978) Ontogeny of synaptic phosphoproteins in brain. Proc Natl Acad Sci USA 75:4037–4041
Luerssen TG, Klauber MR, Marshall LF (1988) Outcome from head injury related to patient’s age. A longitudinal prospective study of adult and pediatric head injury. J Neurosurg 68:409–416
Masliah E, Fagan AM, Terry RD, Deteresa R, Mallory M, Gage FH (1991) Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP-43 in the dentate gyrus of the adult rat. Exp Neurol 113:131–142
Mcphail LT, McBride CB, Mcgraw J, Steeves JD, Tetzlaff W (2004) Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol 185:182–190
Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hubner C, Olney JW, Turski L, Ikonomidou C (1999) N-Methyl-d-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA 96:2508–2513
Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS (1986) Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232:232–235
Rice D, Barone S Jr (2000) Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 108(Suppl 3):511–533
Shojo H, Kibayashi K (2006) Changes in localization of synaptophysin following fluid percussion injury in the rat brain. Brain Res 1078:198–211
Smith ML (1989) Memory disorders associated with temporal lobe lesions. In: Boller F, Grafman J (eds) Handbook of neuropsychology. Elsevier, pp 91–106
Smith TD, Adams MM, Gallagher M, Morrison JH, Rapp PR (2000) Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci 20:6587–6593
Stevenson, KL, Skinner JC, Davis DS, Tran MP, Dixon CE, Kochanek PM, Jenkins LW, Adelson PD (2000) Behavioral dysfunction in immature rats after controlled cortical impact (CCI). J Neurotrauma 17:944
Tran MP, Rodriguez AG, Dixon CE, Kochanek PM, Davis DS, Stevenson KL, Jenkins LW, Adelson PD (2001) Histologic effects of acute NMDA blockade following controlled cortical impact in immature rats. J Neurotrauma 18:1141
Unal-Cevik I, Kilinc M, Gursoy-Ozdemir Y, Gurer G, Dalkara T (2004) Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res 1015:169–174
Uylings HB, Van Eden CG (1990) Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Prog Brain Res 85:31–62
Voigt T, De Lima AD, Beckmann M (1993) Synaptophysin immunohistochemistry reveals inside-out pattern of early synaptogenesis in ferret cerebral cortex. J Comp Neurol 330:48–64
Wiedenmann B, Franke WW (1985) Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41:1017–1028
Acknowledgment
This project was supported by the Walter L. Copeland Fund of The PittsburghFoundation and NIH grant RO1 NS42298-01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gobbel, G.T., Bonfield, C., Carson-Walter, E.B. et al. Diffuse alterations in synaptic protein expression following focal traumatic brain injury in the immature rat. Childs Nerv Syst 23, 1171–1179 (2007). https://doi.org/10.1007/s00381-007-0345-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-007-0345-2