
Abstract
Object
The object was to describe the clinical, radiologic, and pathologic features of astroblastomas in an unselected group of children who were treated in a single institution during an 11-year period.
Methods
Eight children with astroblastomas of the brain were examined. Diagnosis was based on cell morphology, vascular attachment of the cell main process, lack of an epithelial-free surface differentiation, and poor intercellular cohesiveness. In addition to sections, tumor smears and electron microscopy were required for demonstrating or confirming such features.
Conclusions
Clinical findings seem to confirm an apparent predilection of astroblastomas for younger children (median age of onset, 5 years) and the existence of two prognostically different types of tumor—well differentiated (low grade) and anaplastic (high grade). Microscopic findings suggest a closer resemblance of tumor cells to astroblasts rather than to “tanycytes” or ependymal cells. It seems, however, that anaplastic astroblasts have a tendency to evolve toward, or be associated with, less differentiated cells, either neuroepithelial or sarcomatous.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The basic pathologic features of astroblastoma as a distinct neoplasm were delineated early last century by Bailey and associates [2–4]. These workers recognized the presence of cells that closely resembled displaced epithelial cells, or astroblasts. Thus, astroblastoma was initially conceived as a tumor formed by astroblasts, and consequently, recognized by the finding of astroblastic perivascular pseudorosettes. Subsequent authors often paid little attention to the morphology of the cells themselves, and relied mostly on the finding of perivascular pseudorosettes for making the diagnosis [7]. However, without clear recognition of the cells’ distinctive morphology, the distinction of astroblastomas from other tumors, such as ependymomas or astrocytomas, became blurred and controversial. Based on the electron microscopic findings in two cases, a renewed definition of astroblastoma was proposed in 1989 by Rubinstein and Herman [31]. These authors described rudimentary ependymal surface differentiation in such cells, and therefore, suggested that they might represent neoplastic “tanycytes” rather than astroblasts. Tanycytes were thought by Horstmann to be different from previously recognized glial cells [18]. However, neither him nor his followers tried to distinguish tanycytes from the so-called epithelial cells, radial glia, or ependymal cells of earlier authors. Therefore, to what extent tanycytes are different from such cells other than by name remains to be determined. According to the World Health Organization, astroblastoma is classified among neuroepithelial tumors of unknown origin [21]. In practice, however, an ependymal derivation of such cells understandably is suspected by many authors [10].
Findings in the group of patients reported here suggest that tumor cells in astroblastomas are not representative of a surface epithelium, and therefore, that at least in cases like ours, such cells are unlikely to be of an ependymal or tanycytic nature. With the widespread use of magnetic resonance imaging, electron microscopy, and advancement in pathological staining, some progress has been made in the diagnosis of these tumors; nevertheless, its diagnosis remains controversial. In view of the controversy, some of the pathologic features of these tumors will be described in detail. Clinically, it has become apparent that some of these tumors are malignant while in others a more indolent course is to be expected after surgical excision [8]. The importance of such a distinction will be underlined here by the fact that, in patients with malignant tumors, irradiation and chemotherapy seemingly did little to alter the relentless course of the disease.
Materials and methods
This is a retrospective study approved by our Institutional Review Board. Between June 1992 and May 2003, the diagnosis of astroblastoma was made pathologically in eight patients at the Children’s Memorial Hospital. In seven of them, this was the first presentation of the illness and in the remaining patient its sixth recurrence (previous presentations of the tumor in this patient were reported in 1986 by Husain and Leestma) [19]. Pre- and postsurgical computerized tomography (CT) scans or magnetic resonance (MR) images were available in all cases. Gross total resection (GTR) was attempted in all cases. With the exception of patient 6, who was referred for treatment from overseas, the Brain Tumor Clinic at our institution has regularly followed all patients.
At the time of surgery, tumor smears were fixed in alcohol and representative tissue samples were fixed in 10% formalin, B5 fixative, and 2.5% glutaraldehyde. The latter were postfixed in 3% osmium and embedded in epoxi resin. Thin sections were stained with uranyl acetate and lead citrate. Smears were stained with hematoxylin and eosin (H&E). Formalin and B5-fixed tissue samples were embedded in paraffin. Sections were stained with routine histologic methods. Sections from selected formalin and B5-fixed tissue blocks were immunostained by the peroxidase–antiperoxidase method, using antisera against various antigens, including, in every case, GFAP and vimentin.
Results
Clinical findings
All eight of the patients in this series were under the age of 15 at the time of diagnosis. Five of the patients were 5 years old or less at the time of first diagnosis. There were five girls. The mean and median age at the time of diagnosis was 7 years (range 1.8–14.5 years) and 5 years respectively. The mean follow up was 58.5 months (4–184 months; Table 1).
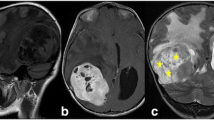
Seven patients were found to have headaches and focal findings and one patient presented with a history of seizures. The location of the tumors was supratentorial in 7 cases and infratentorial in 1. Two of the lesions were located inside the ventricular system: one in the posterior third ventricle and another in the fourth ventricle. In addition, one of the cerebral hemispheric lesions had a small intraventricular component. Radiologically, the tumors were well circumscribed and predominantly solid in 4 cases. A prominent cystic element was apparent in the other 3. In the 6 patients with cerebral hemispheric lesions, 5 of the tumors were located at a distance from the ventricle. Calcification was apparent in 4 patients, mostly in those with a solid lesion. In one of them (patient 7), calcification was massive. All tumors enhanced after contrast infusion both on CT and MR imaging. On T2-weighted MR sequences, most of the lesions were hyperintense with relatively little peritumoral edema. The solid element often had a bubbly appearance, particularly on FLAIR images. T1- and T2-weighted images showed different signal intensities (Fig.I1). The location and radiologic appearance of recurrent tumors usually were similar to those of the original lesions (Table 2).

Radiological findings in patient 7. a, b T1-weighted image shows a hyperintense lesion in the temporal lobe. After gadolinium administration the tumor shows marked enhancement. c On T2-weighted sequences, there is not much perilesional edema and the tumor is slightly hypointense compared with the cortex. d On FLAIR images, it is easier to appreciate the bubbly appearance of this lesion.
Treatment and outcome
Complete surgical resection was attempted in all patients. In patient 5, GTR was limited because the tumor was adjacent to the brainstem. In the event of a tumor recurrence, a resection was re-attempted. However, in patient 2, gamma knife was used because the residual tumor was in close proximity to the brainstem. Following tumor resection, radiation was given to 6 patients. Three of these patients had well differentiated astroblastomas (patients 2, 5, and 8) and 3 patients had anaplastic astroblastomas (patients 1, 4, and 6). Due to young age (1.8 years), a patient with an anaplastic astroblastoma (patient 3) was not irradiated. Five patients received external beam radiation. In addition, 2 patients (6 and 8) received 10 Gy of intraoperative radiation to the tumor bed. In 2 of the patients with well differentiated astroblastomas (patients 2 and 5), external beam radiation was given after surgical excision of the recurrent tumor. In addition, in patient 2, gamma knife was used instead of surgery to treat a second recurrence. Both of these patients have now been free from tumor recurrence for 7 and 5 years respectively (Table 1).
Four patients received chemotherapy as part of their therapeutic regimens. Two patients received chemotherapy following initial surgical resection as part of their initial treatment plan. In patient 4, a regimen of taxol with external beam radiation was followed by maintenance cycles of cyclohexylchloroethylnitrosourea (CCNU), procarbazine, and vincristine. And after recurrence, the child received cisplatin, etoposide, and adriamycin followed by high-dose therapy with thiotepa and cyclophosphamide prior to autologous stem cell rescue. Patient 5 was treated with topotecan prior to second-look and focal radiation therapy. Two other patients received chemotherapy following recurrence. In patient 3, after early recurrence, the tumor was treated with cyclophosphamide, vincristine, VP-16, and cisplatin. Patient 1 had been treated before, following two post-irradiation recurrences with “8 in 1” chemotherapy.
At the time of this report, 4 patients had died (patients 1, 3, 4, and 6)—all with anaplastic astroblastomas. The survival was 15 years, 11 months, 24 months, and 16 months respectively. All patients with well-differentiated astroblastomas are currently alive and disease free with a mean follow-up of 69 months (Table 1).
Pathologic findings
Smears
Characteristic findings included polarization of cells, lack of cohesiveness, and perivascular pseudorosetting. Most tumor cells spread evenly over the slide, rather than in a lumpy manner. They were intermixed with clumps of blood vessels cuffed by tumor cells attached to them like the barbs of a feather (Fig. 2). Cells often seemed polarized, with the nucleus on one end and a tail-like cytoplasmic process on the other (Fig. 3). In some cases, such a process was relatively prominent while in others it was short or inconspicuous. In patients 3 and 8, it was prominent in the first biopsy but inconspicuous in the last. In some cases nuclei varied in size. Binucleation, hyperchromatia, or giant nuclei sometimes were seen.

Patient 3. Perivascular pseudorosette. Blood vessel is cuffed by polarized tumor cells attached to it with their main cytoplasmic process (tumor smear, H&E stain, original magnification, ×400).

Patient 2. Polarization and lack of cohesiveness. In smears tumor cells spread like those of medulloblastomas. Polarization of the nucleus and vascular process is apparent in many cells (tumor smear H&E stain, original magnification, ×650).
Histologic sections
In six cases, perivascular pseudorosettes were immediately obvious in H&E-stained sections as well as in immunopreparations (Fig. 4). In patients 6 and 7, their prominence only was apparent after immunostaining for vimentin or GFAP. Due to cell looseness in the intervascular region, pseudopapillae often were formed (Fig. 5). In most cases, reactivity for vimentin was stronger and more consistent than for GFAP. In patient 8, only some of the perivascular processes were reactive for either one.

Patient 2. Polarization and perivascular pseudorosettes. Tumor cells are monopolar, with their nucleus in one end of the cell and their main or vascular process in the other (vimentin immunostain, original magnification, ×650).

Patient 2. Pseudopapillae. Except for their attachment to blood vessels, tumor cells exhibit little intercellular cohesiveness and tend to spread apart from each other. Thus, in areas like this perivascular pseudorosettes are seen as pseudopapillae (phosphotungstic acid-hematoxylin [PTAH] stain, original magnification, ×200).
In patient 1, this was its sixth recurrence after a remission of 9 years. There was extensive hyalinization of the stroma. In a few areas, true rosettes as well as ribbon-like cords of columnar hyperchromatic cells were found (Fig. 6). Such unexpected structures were reminiscent, but not diagnostic, of medulloepithelioma. In patient 4, the tumor was anaplastic and an exhuberant proliferation of blood vessels was seen within the cores of many pseudorosettes (Fig. 7). Following complete excision, local recurrence of the lesion 1 year later disclosed replacement of the original tumor by rhabdomyosarcoma. No astroblastoma vestiges were found. The patient expired from his illness after surviving for 1 more year. No autopsy was performed. In patient 6, a large frontal lobe tumor was biopsied overseas and referred to us for surgical excision. In addition to the astroblastoma, a smaller portion of the tumor had the appearance of an anaplastic astrocytoma (Fig. 8). In patient 7, extensive calcification required decalcification of some tumor blocks.

Patient 1. Primitive rosettes. Monolayered epithelial rosettes with sharply demarcated luminal and external or mesenchymal cell margins are seen H(H&E stain, original magnification, ×400).

Patient 4. Anaplastic astroblastoma. Blood vessels are markedly hyperplastic within the core of some pseudorosettes (Masson’s trichrome stain, original magnification, ×400).

Patient 2. Cell polarization is evident (compared with their appearance in smears, see Fig. 2). Cells are loose and free of junctional complexes. Tips of the perivascular processes are coated with basement membrane (uranyl acetate and lead citrate, original magnification, ×3,000).
Ultrastructure
While perivascular pseudorosettes were present in all cases, ependymal microrosettes were not seen in any of them. Small, non-rosettal microvilli or microvilli-like processes were present in some cells. In patient 8, such processes were seen throughout the perimetry of many cells and often contained microtubules. The main cytoplasmic processes were oriented toward blood vessels. Some were short and stout, others narrow and tapered. Their tips were coated with basement membrane. In patients 2 and 8, some of these processes had terminal knobs filled with mitochondria and vesicles (Fig. 8). Intermediate filaments were consistently present, but sometimes limited to some of the perivascular processes. Microtubules were not as common, except in patient 8. Nuclei were ovoid in some cases and elongated or irregular in others. Nucleoli often were prominent. In patient 3, some cells were lipidized. In the first biopsy from patient 4, many cells contained clusters of dark lysosomes. In its final biopsy the tumor was completely different: small as well as multinucleate giant cells with irregular nucleus, large nucleolus, multiple fragments of immature myofibrils, and patchy basement membrane were seen within a collagenous stroma.
Discussion
Astroblastoma is a rarely diagnosed tumor. Not a single instance of the neoplasm was identified in a review of H&E-stained slides from 3,291 cases of childhood central nervous system tumors that were collected over a period of decades in ten North American institutions [13]. On the other hand, using a silver carbonate method Scharenberg and Liss made the diagnosis of astroblastoma in 9 out of 2,000 cases of gliomas studied during a 37-year period [33]. In their original study, Bailey and Bucy described the lesion mostly in adults [3]. Only 2 of their 25 patients were first diagnosed before the age of 21. However, in series based on consultation material from varied sources, children frequently have been affected [8, 9, 35]. The incidence of astroblastoma in the literature has been estimated to be between 0.45 and 2.8% [24, 33]. In our series, astroblastoma represented 0.92% of the tumors in our pediatric database. In our exclusively pediatric material, 5 out of 8 patients were 5 years old or less at the time of diagnosis. In some of the reported series with a predominance of childhood cases, the number of affected girls has been much larger than that of boys [9, 34]. Although some authors believe that there is no sex predominance, our review of the literature shows that 64% of the patients were female (Table 3) [3, 8, 32]. Our series also had a female predominance of 63%.
The present findings support Bonnin and Rubinstein’s division of astroblastomas into two categories—low grade (well differentiated) and high grade (anaplastic)—with apparently different prognoses [8]. As pointed out by these authors, however, histologic determination of malignancy is not always as clear cut as one would like it to be. Thus, in some cases it is not a simple matter of looking for the presence or absence of anaplastic features—such as atypia, mitoses, or necroses—but of evaluating their degree and making an empirical diagnosis of malignancy based on the overall pattern of the lesion. In our material, cases could be segregated into one of three different categories:
-
1.
In patients 2, 5, 7, and 8, neither in primary nor recurrent tumors was there any evidence of anaplasia other than nuclear atypia in cells with well-formed vascular processes
-
2.
In patients 3, 4, and 6, primary tumors looked malignant due to invasiveness, high cellularity, necroses, and a frequently disorganized pattern of the lesions; furthermore, in two of these cases histologically different malignant neoplasms were closely associated with the primary or recurrent tumors
-
3.
In patient 1, mitoses and necroses were not uncommon in some of the earlier, well-circumscribed and non-invasive recurrences of the neoplasm (at one time the lesion was thought by another pathologist to represent a primitive neuro-ectodermal tumor)
In categories 1 and 2, clinical and pathologic findings were concordant. In the single case in category 3, however, in spite of its histologic appearance, the tumor behaved for many years like a benign lesion. In Bonnin and Rubinstein’s material, well-differentiated astroblastomas seemed more common in childhood [8]. However, in our series, anaplastic and well-differentiated astroblastomas were found in equal numbers.
Based on the present findings, it is possible to say that astroblastoma is a tumor formed by relatively well-differentiated glial cells, which:
-
1.
Are polarized and monopolar
-
2.
Have a single or main cytoplasmic process attached to a blood vessel
-
3.
Lack a free epithelial surface differentiation
-
4.
Are poorly cohesive among themselves
-
5.
Often have their perikaryon and processes richly endowed with vimentin or glial intermediate filaments
Furthermore, because of these features, such tumors are characterized histologically by:
-
6.
Formation of perivascular pseudorosettes
-
7.
Absence of true rosettes
-
8.
Looseness of their architecture, often with a tendency to form pseudopapillae
Most of these features were described by Bailey and Bucy, who emphasized the astroblastic appearance of the cells, the absence of ependymal rosettes and blepharoplasts, and the lack of cohesiveness leading to the formation of pseudopapillae [3]. Additional features of the tumor include:
-
9.
A predominance of cerebral hemispheric lesions
-
10.
A peripheral rather than periventricular location
-
11.
A nodular, non-invasive type of growth
-
12.
A rich vasculature
-
13.
A tendency to perivascular as well as vascular fibrosis and hyalinization
-
14.
A relative lack of local spread or metastases
Some of these latter features were emphasized by Bonnin and Rubinstein and have been adopted for diagnosis by other authors [8–10]. It should be noted, however, that in a given case, these findings are not as reliable for diagnosis as the purely microscopic features of the tumor.
Although cerebral hemispheric lesions are far more common, involvement of infratentorial structures does not seem out of proportion with the relative amount of normal brain tissue in that compartment. Two of Bonnin and Rubinstein’s 23 cases and one of Scharenberg and Liss’ 9 patients had the tumor in the posterior fossa [8, 33]. Similarly, in 1 of our 8 patients the tumor was in the fourth ventricle. While cerebral astroblastomas frequently develop at a distance from the ventricle, periventricular, and intraventricular lesions can be found, as demonstrated by patients 2, 5, and 6. Astroblastomas often are relatively well circumscribed and seem to grow mainly by expansion rather than infiltration. However, such a growth pattern does not distinguish them from hemispheric cerebral ependymomas.
In three of our patients, a seemingly different neoplasm developed in close association with the original tumor. In patient 6, part of the tumor was astrocytic at the time of presentation. In patient 1, less differentiated, medullo-epithelioma-like elements were found within the recurrent tumor 14 years after its original presentation. In patient 4, 1 year after its first excision the lesion recurred as a rhabdomyosarcoma. While the association of astroblastoma with astrocytoma was recognized many years ago, to our knowledge development of medullo-epithelioma or rhabdomyosarcoma in such a tumor has not been previously documented [14]. Exceptionally, rhabdomyosarcoma has been seen in association with glioblastomas, known as the gliomyosarcoma [6, 16]. On the other hand, striated muscle sometimes is seen in cerebral medullo-epithelioma, or more commonly, in ciliary teratoid medullo-epithelioma [1, 27]. Although the relevance of such findings in cases of astroblastoma remains uncertain, it should be noted that glioblastomas and, occasionally, gliosarcomas have already been documented as complications of astroblastomas in childhood [8]. In any event, in view of the rarity of astroblastomas, our findings suggest that the cell line in such a neoplasm might be relatively unstable.
As seen in our cases, tumor smears fixed in alcohol and stained with H&E often demonstrate three of the main features of astroblastoma cells:
-
1.
Their polarized monopolar morphology
-
2.
Their rosetting around blood vessels
-
3.
Their lack of cohesiveness
To some extent, ependymoma cells sometimes share the first two of these features; nevertheless, they are rather cohesive and tend to aggregate in lumps in a manner similar to that of astrocytomas. As in our patient 3, smears of anaplastic astroblastoma can sometimes resemble those of medulloblastoma, with naked hyperchromatic nuclei and barely visible perikarya and cell processes. In medulloblastoma smears, however, no perivascular pseudorosettes are found.
As in the majority of gliomas, the immunohistochemical profile in astroblastomas is not very specific. Nevertheless, although immunoreactivity per se might not be diagnostic of any given glioma, immunohistochemistry is most useful for delineating in sections the shape of neoplastic glial cells. Thus, while reactivity, say, for vimentin in a glioma would not mean much per se, such a preparation might be diagnostic if it succeeds in outlining the cell processes. In our experience, immunohistochemistry in B5-fixed specimens has been far superior than in formalin-fixed material for demonstrating cell morphology in neural tissue.
The finding of papillae has been emphasized in some reports of astroblastomas [11]. However, from the published illustrations it would appear that, as in our cases, such structures result from tissue breakdown, not from papillary growth. Therefore, we would rather refer to them as pseudopapillae. In our opinion, the presence of true papillae, and thus of a free epithelial surface, would be inconsistent with the diagnosis of astroblastoma. It is precisely for this reason that electron microscopy is needed to verify the absence of such an epithelial surface before making the diagnosis of this particular tumor.
The main contribution of electron microscopy in our cases was to verify the absence of a free epithelial surface differentiation within the tumor: in other words, to rule out the possibility of ependymoma. While the ultrastructural diagnosis of a well-differentiated ependymoma is relatively simple, electron microscopic distinction between an astroblastoma and an anaplastic ependymoma can be rather difficult. The problem being that in anaplastic ependymomas tumor cells tend to lose, or not to develop, some of their epithelial surface features. Thus, microrosettes are small and rudimentary, with poorly developed junctional complexes and often without cilia.
Light microscopic as well as ultrastructural findings in our cases would support Bailey and Bucy’s interpretation of the tumor cells of astroblastoma as being displaced epithelial cells or astroblasts rather than ependymal tanycytes. A few points made by Ramón y Cajal may still be relevant for the purpose of this discussion [30]. Astrocytes like those seen in the mature human brain are mostly characteristic of birds and mammals. For the most part, early in human development and in mature lower vertebrates glial cells remain a surface epithelium. Such primitive ependymal or epithelial cells do have a peripheral process that extends to the external brain surface, either at its interface with the pia mater or at the developing perivascular space. With development, some epithelial cells lose their peripheral process and become mature ependymal cells. Conversely, other epithelial cells migrate, lose their epithelial features, and become astroblasts. Astroblasts, therefore, are by definition displaced epithelial cells that are removed from the ventricle and have lost their free surface epithelial features, including their intercellular cohesiveness. However, they still are polarized, conserve their peripheral process, and have not yet developed the dendritic features of an astrocyte.
On the other hand, it should be noted that ordinary ependymomas are characterized by a rarely emphasized feature: the alignment of tumor cells on a free epithelial surface. In most cases such a surface is limited to microrosettes and, although present everywhere, is not readily visible under the light microscope. It should also be realized that most cells in ordinary ependymomas resemble primitive rather than mature ependymal cells. In fact, such tanycytic-like primitive ependymal cells commonly are recognized by their peripheral processes making contact with blood vessels in a rosette-like manner. The more mature—and less prevalent—cells in those tumors tend to form ependymal rosettes and to lose their peripheral processes.
Astroblastomas are usually located in the cerebral hemispheres. However, other locations such as the corpus callosum, cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem have been reported [5, 8, 19, 33]. In our cases, we had two tumors with locations not in the cerebral hemispheres: one in the fourth ventricle and another in the posterior third ventricle. Although these tumors are intra-axial in nature, there are some reports that claim their extra-axial appearance [5, 37]. Most of the astroblastomas are a combination of cystic and solid components. The solid portion of these lesions usually has a “bubbly” appearance, probably related to the angioarchitecture of these tumors where the vessels make up the supporting tissue for astroblasts. This is seen as punctate signal voids on MRI [5, 15]. In addition, the presence of microcalcifications can also contribute to the look of these neoplasms. Astroblastomas tend to be well demarcated and seem to grow mainly by expansion; nevertheless, infiltrating tumors have been reported, especially in cases of high-grade tumors [17]. These tumors have intense heterogeneous enhancement after contrast administration, both on CT and MRI. The MRI appearance is compatible with other glial tumors, with variable intensities both on T1- and T2-weighted images. Port et al. suggested that there are some specific features that can help differentiate astroblastomas from other neoplasms, especially ependymomas [29]. According to these authors, the tumors have relatively little peritumoral edema for their size, the cystic component is usually complex, the solid component has the characteristic bubbly look, and they have low signal intensity on T2-weighted images [29], although in our experience, only one of the tumors was relatively hypointense on T2-weighted images.
Due to the small number of astroblastoma cases reported in the literature, there is no consensus as to the biologic behavior. A WHO grade for astroblastomas has not been established. However, differentiating astroblastomas are considered to have a better prognosis than anaplastic astroblastomas [8, 35]. While GTR is the best therapeutic option, adjuvant therapy after tumor removal can be considered in patients with anaplastic astroblastomas. We believe that patients with well-differentiated astroblastomas should be observed, reserving future treatments for any recurrence. Because recurrence is usually local, we recommend further resection and adjuvant treatment. However, Thiessen at al. and Bonnin and Rubinstein have described distant spread of the disease [8, 35]. In addition, a well-differentiated lesion that recurred in the pineal region was treated with external beam on first recurrence and gamma knife on second recurrence. Neither intra-operative radiation nor gamma knife has been previously reported for the treatment of astroblastomas. Several authors have also opted for radiation therapy as a first option for adjuvant therapy in astroblastomas [5, 8, 9]. However, adjuvant therapy needs to be further explored before its effectiveness can be determined.
In our series four children received a variety of chemotherapeutic regimens. These regimens were used based both on the pathologic diagnosis and the patient’s age. Given the small number of children in this series and the variety of regimens used no conclusions can be drawn as to the usefulness of chemotherapy for the treatment of astroblastomas.
Conclusion
An astroblastoma, although rare, has to be considered in the differential diagnosis of tumors in childhood, and especially in the differential diagnosis of an ependymoma. Its resemblance to this tumor makes the diagnosis of an astroblastoma challenging. In addition, we have found that this tumor can be pathologically unstable. Most of the cases are supratentorial with a slight female predominance. The best treatment modality is surgical resection. The prognosis for well-differentiated lesions is similar to that for low-grade gliomas. In those with anaplastic features, adjuvant therapy (radiotherapy and/or chemotherapy) upfront can be considered since their behavior and prognoses resemble those of anaplastic astrocytomas.
References
Auer RN, Becker LE (1983) Cerebral medulloepithelioma with bone, cartilage, and striated muscle. Light microscopic and immunohistochemical study. J Neuropathol Exp Neurol 42:256–267
Bailey P (1965) Cellular types in primary tumors of the brain. In: Penfiled W (ed) Cytology and cellular pathology of the nervous system. Hoeber, New York, p 1932. Reprinted by Hafner, New York
Bailey P, Bucy PC (1930) Astroblastomas of the brain. Acta Psychiatr Neurol 5:439–461
Bailey P, Cushing H (1926) A classification of the tumors of the glioma group on a histogenic basis with a correlation study of prognosis. Lippincott, Philadelphia
Baka JJ, Patel SC, Roebuck JR, Hearshen DO (1993) Predominantly extraaxial astroblastoma: imaging and proton MR spectroscopy features. Am J Neuroradiol 14:946–950
Barnard RO, Bradford R, Scott T, Thomas DGT (1986) Gliomyosarcoma. Report of a case of rhabdomyosarcoma arising in a malignant glioma. Acta Neuropathol (Berl) 69:23–27
Bergstrand H (1935) Über das sog. Astrocytom in Kleinhirn. Virchows Arch A Pathol Anat Histopathol 287:538–548
Bonnin JM, Rubinstein LJ (1989) Astroblastomas a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery 25:6–13
Brat DJ, Hirose Y, Cohen KJ, Feuerstein BG, Burger PC (2000) Astroblastoma: clinicopathologic features and chromosomal abnormalities defined by comparative genomic hybridization. Brain Pathol 10:342–352
Burger PC, Scheithauer BW (1994) Tumors of the central nervous system. In: Atlas of tumor pathology, third series, fascicle 10. Armed Forces Institute of Pathology, Washington, DC
Cabello A, Madero S, Castresana A, Diaz-Lobato R (1991) Astroblastoma: electron microscopy and immunohistochemical findings: case report. Surg Neurol 35:116–121
Catalan-Uribarrena G, De Las Heras-Echeverria P, Caton-Santaren B, Martinez-Soto LJ, Torrecilla-Sardony MV, Ramos-Gonzalez A (2002) Cerebral astroblastoma: report of a case and literature review. Neurocirugia 13:378–384
Childhood Brain Tumor Consortium (1998) A study of childhood brain tumors based on surgical biopsies from ten North American institutions: sample description. J Neurooncol 6:9–23
Del Río-Hortega P (1962) Anatomía microscópica de los tumores del sistema nervioso central y periférico. In: Proceedings of the Congreso Internacional de Lucha Científica y Social Contra el Cáncer. Madrid, 1933. English translation by A Pineda, GV Russell, KM Earle. Thomas, Springfield
De Reuck J, Van de Velde E, Vander Eecken H (1975) The angioarchitecture of the astroblastoma. Clin Neurol Neurosurg 78:89–98
Goldman RL (1969) Gliomyosarcoma of the cerebrum. Report of a unique case. Am J Clin Pathol 52:741–744
Hoag G, Sima AA, Rozdilsky B (1986) Astroblastoma revisited: a report of three. Acta Neuropathol 70:10–16
Horstmann E (1954) Die Faserglia des Selachiergehirns. Z Zellforsch Mikrosk Anat 39:588–617
Husain AN, Leestma JE (1986) Cerebral astroblastoma: immunohistochemical and ultrastructural features. Case report. J Neurosurg 64:657–661
Jay V, Edwards V, Squire J, Rutka J (1993) Astroblastoma: report of a case with ultrastructural, cell kinetic, and cytogenetic analysis. Pediatr Pathol 13:323–332
Kleinhius P, Burger PC, Scheithauer BW (1993) Histologic typing of tumors of the central nervous system. In: World Health Organization, international histological classification of tumors. Springer, Berlin Heidelberg New York
Kubota T, Hirano A, Sato K, Yamamoto S (1985) The fine structure of astroblastoma. Cancer 55:745–750
Kujas M, Faillot T, Roncier B, Catala M, Poirier J (2000) Astroblastoma revisited. Report of two cases with immunohistochemical and electron microscopic study. Histogenic considerations. Neuropathol Appl Neurobiol 26:295–298
Kuroiwa T, Ito U, Yamaguchi T, Okada K, Inaba Y (1980) [A case of astroblastoma of cerebral hemisphere in infant (Kobe)] (in Japanese). Shoni No Noshinkei 5:107–112
Mierau GW, Tyson RW, McGavran L, Parker NB, Partington MD (1999) Astroblastoma: ultrastructural observations on a case of high-grade type. Ultrastruct Pathol 23:325–332
Nakajima F, Yuda K, Kuwabara T, Yagishita S (1982) [Astroblastoma in newborn baby] (in Japanese). Neurol Surg 10:291–293
Nicholson DH, Green WR (1981) Pediatric ocular tumors. Masson, New York
Pizer B, Moss T, Oakhill A, Webb D, Coakham H (1985) Congenital astroblastoma: an immunohistochemical study. Case report. J Neurosurg 83:550–555
Port J, Brat D, Burger P, Pomper M (2002) Astroblastoma: radiologic–pathologic correlation and distinction from ependymoma. Am J Neuroradiol 23:243–247
Ramón y Cajal S (1952) Histologie du système nerveux de l’homme et des vertébrés. Translated by L. Azoulay. Maloine, Paris, 1909. Reprinted by Consejo Superior de Investigaciones Científicas, Madrid
Rubinstein LJ, Herman MM (1989) The astroblastoma and its possible cytogenetic relationship to the tanycyte. An electron microscopic, immunohistochemical, tissue- and organ-culture study. Acta Neuropathol 78:479–483
Sano K (1969) [Astrocytoma and astroblastoma] (in Japanese). No To Shinkei 21:610–613
Scharenberg K, Liss L (1969) Neuro-ectodermal tumors of the central and peripheral nervous system. Williams and Wilkins, Baltimore, pp 17–29
Shuangshoti S, Mitphraphan W, Kanvisetsri S, Griffiths L, Navalitloha Y, Pornthanakasem W, Mutirangura A (2000) Astroblastoma: report of a case with microsatellite analysis. Neuropathology 20:228–232
Thiessen B, Finlay J, Kulkarni R, Rosenblum MK (1998) Astroblastoma: does histology predict biologic behavior? J Neurooncol 40:59–65
Yamashita J, Handa H, Yamagami T, Haebara H (1985) Astroblastoma of pure type. Surg Neurol 24:218–222
Yunten N, Ersahin Y, Demirtas E, Yalman O, Sener RN (1996) Cerebral astroblastoma resembling an extra-axial neoplasm. J Neuroradiol 23:38–40
Acknowledgements
We are grateful to Ms. D. Sciortino for printing the photomicrographs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Navarro, R., Reitman, A.J., de León, G.A. et al. Astroblastoma in childhood: pathological and clinical analysis. Childs Nerv Syst 21, 211–220 (2005). https://doi.org/10.1007/s00381-004-1055-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-004-1055-7