
Abstract
Several previous studies indicated that Actinobacteria may be enriched in soils with elevated content of heavy metals. In this study, we have developed a method for the in-depth analysis of actinobacterial communities in soil through phylum-targeted high-throughput sequencing and used it to address this question and examine the community composition in grassland soils along a gradient of heavy metal contamination (Cu, Zn, Cd, Pb). The use of the 16Sact111r primer specific for Actinobacteria resulted in a dataset obtained by pyrosequencing where over 98 % of the sequences belonged to Actinobacteria. The diversity within the Actinobacterial community was not affected by the heavy metals, but the contamination was the most important factor affecting community composition. The most significant changes in community composition were due to the content of Cu and Pb, while the effects of Zn and Cd were relatively minor. For the most abundant actinobacterial taxa, the abundance of taxa identified as members of the genera Acidothermus, Streptomyces, Pseudonocardia, Janibacter and Microlunatus increased with increasing metal content, while those belonging to Jatrophihabitans and Actinoallomurus decreased. The genus Ilumatobacter contained operational taxonomic units (OTUs) that responded to heavy metals both positively and negatively. This study also confirmed that Actinobacteria appear to be less affected by heavy metals than other bacteria. Because several Actinobacteria were also identified in playing a significant role in cellulose and lignocellulose decomposition in soil, they potentially represent important decomposers of organic matter in such environments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recent reports show that in addition to fungi, also bacteria play a significant role in cellulose and lignocellulose decomposition in soil environments (Baldrian et al. 2012; Berlemont and Martiny 2013). Among bacteria, the members of the phylum Actinobacteria show the highest abundance of genes involved in cellulose decomposition in their genomes (Berlemont and Martiny 2013), and many isolates have been shown to be able to efficiently degrade polysaccharides and polyphenols (Abdulla and El-Shatoury 2007; Anderson et al. 2012; Chater et al. 2010; Enkhbaatar et al. 2012; Větrovský et al. 2014; Yin et al. 2010). Bulk lignocellulose materials are more accessible for decomposition by filamentous microorganisms such as fungi and Actinobacteria (de Boer et al. 2005), and efficient growth on straw was indeed demonstrated for environmental isolates of Actinobacteria (Větrovský et al. 2014). Filamentous mycelial growth, the ability to form spores and a powerful secondary metabolism also represent suitable traits for the survival of Actinobacteria under stress conditions such as heavy metal-contaminated soils, for which these bacteria have also developed several resistance mechanisms (Bajkic et al. 2013; Ivshina et al. 2013; Schmidt et al. 2005). This unique combination of traits makes Actinobacteria good candidates for the efficient decomposition of lignocellulose in heavy metal-contaminated areas where they may potentially represent the major metabolically active group. While the abundance of other bacteria phyla typically decreases with increasing heavy metal bioavailability, previous studies demonstrated that the abundance of the Actinobacteria is similar or higher than in clean soils of similar properties (Berg et al. 2012; Gremion et al. 2003).
Recent high-throughput sequencing-based studies of bacterial communities in soil revealed that Actinobacteria can be highly abundant in certain soils, such as those of arid ecosystems, where they often represent over 45 % of the bacterial community (Bachar et al. 2010; Dunbar et al. 1999), while in typical soils, they represent only between <5 and 20 % of the bacterial community (Baldrian et al. 2012; Dominguez-Mendoza et al. 2014; Lauber et al. 2009). Because of the importance of Actinobacteria in soil processes and their varying abundance, previous attempts have been made to analyse the composition of their communities by phylum-targeted approaches. Studies published so far have unfortunately been limited to fingerprinting methods, such as DGGE and T-RFLP, in combination with other molecular identification techniques such as clone sequencing (Liu et al. 2013; Steger et al. 2007; Wang et al. 2013) and do not offer sufficient depth for a detailed analysis of the composition of their communities in soils. However, the massive parallel sequencing that is currently available promises to overcome this limitation.
Through massive parallel sequencing, the aims of this study were to develop a method for an in-depth analysis of actinobacterial communities in soil and to provide an effective tool for the detailed examination of their community composition. This approach was used to describe the actinobacterial community in grassland soils developed at a gradient of heavy metal contamination from the vicinity of a polymetallic smelter. The study sites for this study were selected to represent a long-term gradient of total heavy metal content (Cu, Zn, Cd, Zn, Pb and As) which was due to the variable distance from a local polymetallic smelter. Although the available concentrations of heavy metals may seem to be more relevant for studying biological effects, we have decided to base the analyses on the total heavy metal content for several reasons: (i) the metal availability can be variable in time both short-term and long-term and there is not sufficient information on the history of heavy metal availability in the area; (ii) availability of heavy metals is highly affected by the method used for extraction, for example, in the area of study, two routinely used methods delivered estimates differing almost by one order of magnitude (Mühlbachová et al. 2015), and this makes comparisons to other studies where heavy metal availability was estimated difficult; and (iii) for the area of this study, the correlations of total and available heavy metal contents were found to be both linear and highly significant. Also, the effects of total and available heavy metals on soil functions (respiration, dehydrogenase activity) were both significant (Mühlbachová et al. 2015).
The analysis was aimed to verify the hypothesis that the relative abundance of Actinobacteria increases with the increasing heavy metal availability and to identify the effects of heavy metal content on the diversity and composition of the actinobacterial community. We hypothesised that the increasing heavy metal toxicity only allows the existence of a limited number of resistant actinobacterial taxa, decreasing their overall diversity in soil. This can be potentially of critical importance for soil functioning: several actinobacterial taxa from the area of this study were previously demonstrated to decompose multiple polysaccharides and phenolics and to grow on complex lignocellulose compounds (Větrovský et al. 2014). This indicates their involvement in the decomposition of plant-derived biomass in the studied soil which can be potentially harmed by heavy metal toxicity.
Materials and methods
Study site, soil sample collection and analysis
The study area was located near Příbram, Czech Republic (49° 42′ 22.207″ N, 13° 58′ 27.296″ E). The area was covered by a mixed grassland. The soil was a Cambisol with a pH ranging from 5.5 to 6.5 and a clay/silt/sand ratio of approximately 40:30:30 %. The smelter was in operation from 1786, and it originally mostly worked lead ores. Metal mining ceased in 1972, but the smelter still processes secondary lead and cadmium sources. The present levels of heavy metal contamination are reported from the soils over the period >30 years which makes this area suitable for the exploration of long-term heavy metal contamination (Kalac et al. 1991; Mühlbachová 2011; Mühlbachová et al. 2015).
The sampling was performed in June 2012 and November 2012 to cover potential seasonal differences in community composition. The same five sites, located along a gradient of contamination represented by distances of 300 to 2500 m from the polymetallic smelter, were sampled on both occasions.
At each site, four samples were collected using plastic cores of a 4.5-cm diameter. The samples were transported to the laboratory and stored at 4 °C until processing, which was performed within 24 h. The top 5 cm of each soil core were collected, the roots and stones were removed, and the soil was sieved through a 5-mm sterile sieve and mixed. Sieved samples were freeze-dried and kept at −40 °C until analysis.
DNA was extracted from 300 mg of freeze-dried soil of each sample using the SV method (Sagova-Mareckova et al. 2008) and cleaned using the GeneClean Turbo Kit (MP Biomedicals, Solon, OH, USA). The sample pH was measured in soil water extract (1 g soil/10 ml deionised water), and the soil moisture content was calculated by estimating the soil dry mass before and after freeze-drying. Oxidizable C (Cox) and total N (Ntot) content was measured using an elemental analyser in an external laboratory (Research Institute for Soil and Water Conservation, Prague, Czech Republic). Cox was measured using sulfochromic oxidation (ISO 14235), and nitrogen content was estimated by sulphuric acid mineralisation with the addition of selenium and sodium sulphate and conversion to ammonium ions (ISO 11261), which were measured by the segmented flow analyser (SFA) Skalar. The content of heavy metals (Cd, Cu, Pb, Zn) was measured after decomposition by aqua regia on the atomic absorption spectrometer Varian 240.
Quantification of microbial biomass
Two sets of specific PCR primers were used to quantify the relative amounts of actinobacterial and bacterial DNA: Actino235 (CGCGGCCTATCAGCTTGTTG) (Stach et al. 2003) and Eub518 (ATTACCGCGGCTGCTGG) (Muyzer et al. 1993) for Actinobacteria and 1108 F (ATGGYTGTCGTCAGCTCGTG) (Amann et al. 1995) and 1132R (GGGTTGCGCTCGTTGC) (Wilmotte et al. 1993) for bacteria. Real-time PCR assays were based on the method of Leigh et al. (Leigh et al. 2007). Amplifications were performed on a Step One Plus cycler (Applied Biosystems) using optical grade 96-well plates. Each 15 μl reaction mixture contained 7.5 μl SYBR Green Master Mix (Applied Biosystems), 0.6 μl BSA (10 mg/ml), 0.9 μl of each primer, 1.0 μl of template and 4.1 μl of water. The PCR cycling protocol was the same for actinobacterial and bacterial DNA quantification: incubation at 56 °C for 2 min and at 95 °C for 10 min, followed by 40 cycles of amplification consisting of 15 s at 95 °C and 1 min at 60 °C. Cloned fragments of the 16S rRNA gene from Streptomyces lincolnensis DNS 40335 were used as standards.
Tag-encoded amplicon pyrosequencing of actinobacterial community
The combination of a universal primer, eub530F (GTGCCAGCMGCNGCGG (Dowd et al. 2008)) and an Actinobacteria-specific primer 16Sact1114r (GAGTTGACCCCGGCRGT, (Kyselkova et al. 2008)) was used to selectively amplify partial 16S sequences of the Actinobacteria. As a proxy of primer coverage of the Actinobacteria-specific primer (i.e. the percentage of actinobacterial sequences in a dataset from which the primer set generates amplicons) and specificity (i.e. the percentage of non-actinobacterial sequences amplified by the primers in other bacterial phyla), the percentages of sequences with primer hits with zero to three mismatches was tested in silico on a 16S rRNA dataset derived from RDP and GenBank covering the primer region (185,400 sequences belonging to 2316 genera), allowing up to three mismatches.
Tag-encoded samples for 454 pyrosequencing were obtained in two steps. In the first step, three PCR reactions were performed independently for each extracted DNA sample using the above primer pair to selectively amplify part of the 16S rRNA gene from Actinobacteria. Each 25 μl reaction mixture contained 16.75 μl H2O, 2.5 μl 10× buffer for DyNAzyme II DNA polymerase, 1.5 μl 10 mg/ml BSA, 1 μl forward primer (final concentration 10 pmol/μl), 1 μl reverse primer (final concentration 10 pmol/μl), 1 μl template DNA, 0.75 μl 4 % Pfu polymerase/DyNAzyme DNA Polymerase (final concentration 2 U/μl) and 0.5 μl PCR Nucleotide Mix (10 mM). The cycling parameters were 5 min at 94 °C, 35 cycles of 60 s at 94 °C, 60 s at 57 °C and 60 s at 72 °C, followed by 10 min final extension at 72 °C. The size and quality of the PCR products were verified on an agarose gel. The three primary PCR reactions from each sample were pooled, cleaned using the MinElute PCR Purification kit (Qiagen, Hilden, Germany) and concentrated to 20 μl.
Secondary PCR was performed using composite eub530f/eub1100br primers modified from Dowd et al. (2008) as described previously (Baldrian et al. 2012). Each 50 μl reaction mixture contained 38.2 μl H2O, 5 μl 10× buffer for DyNAzyme II DNA polymerase, 1.5 μl DMSO, 0.4 μl tagged forward primer (final concentration, 10 pmol/μl), 0.4 μl reverse primer (final concentration, 10 pmol/μl), 2 μl template DNA, 1.5 μl 4 % Pfu polymerase/DyNAzyme DNA Polymerase (final concentration, 2 U/μl) and 1 μl PCR Nucleotide Mix (10 mM). Cycling parameters were 5 min at 94 °C, 15 cycles of 60 s at 94 °C, 60 s at 64 °C, 60 s at 72 °C, followed by 10 min final extension at 72 °C. PCR products were checked on an agarose gel and purified using AMPure Beads (Agencourt, Beverly, MA, USA) to remove short fragments. The concentration of the purified PCR product was quantified using the Quant–iT™ PicoGreen ds DNA kit (Invitrogen, Grand Island, NY, USA), and an equimolar mixture of all samples was prepared. The mixture was subjected to electrophoresis, and after separation, the band with appropriate size was excised, purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA), denatured (2 min. 95 °C) and again cleaned using AMPure Beads. This product was cleaned using the MinElute PCR Purification kit (Qiagen, Hilden, Germany) and quantified by the Kapa Library Quantification kit (Kapa Biosystems, Woburn, MA, USA). The 454 pyrosequencing was performed on the GS Junior instrument (Roche, Basel, Switzerland).
Bioinformatic analysis
Sequencing data were filtered and trimmed using the pipeline SEED v 1.2.1 (Větrovský and Baldrian 2013a). All of the sequences with mismatches in tags were removed from the dataset. Pyrosequencing noise reduction was performed using the PyroNoise algorithm translation within Mothur v 1.28.0 (Schloss et al. 2009); chimeric sequences were detected using the Uchime implementation in USEARCH v 7.0.1090 (Edgar et al. 2011) and deleted. All sequences were trimmed to 380 bp and clustered into operational taxonomic units (OTUs) using UPARSE implementation in USEARCH 7.0.1090 (Edgar 2013) with a 97 % similarity threshold. The consensus from each OTU was constructed from an MAFFT alignment (Katoh et al. 2009) based on the most abundant nucleotide at each position. For sample comparison and diversity analyses, non-actinobacterial sequences were removed, and the dataset was randomly resampled at the same sampling depth of 400 sequences per sample. The sequence data were deposited in the MG-RAST public database (http://metagenomics.anl.gov/, dataset numbers 4638524.3, 4638526.3, 4638527.3, 4638521.3, 4638519.3, 4638523.3, 4638518.3, 4638525.3, 4638522.3, 4638520.3).
OTU identification, phylogenetic analyses and statistics
The identification of OTUs was performed by BLASTn against a local database derived from the Ribosomal Database Project (Cole et al. 2005) from 25.2.2014. Each OTU was assigned to the taxonomic level of genus (or nearest lower level), comparing the BLASTn best hits with the taxonomic information from the NCBI taxonomy server. A dataset containing only actinobacterial sequences was used for OTU table construction. To reduce the bias caused by different numbers of rRNA gene copies in bacterial genomes, read abundances of each OTU were divided by the copy number of the rRNA genes in the genome of the closest taxon with a complete genome sequence as described previously (Větrovský and Baldrian 2013b).
Phylogenetic analysis and tree construction was performed in MEGA 6 (Tamura et al. 2013), and a circular graphical tree representation was prepared using the program GraPhlAn (http://huttenhower.sph.harvard.edu/graphlan). Statistical tests and principal component analysis were conducted using the software package Statistica 7 (StatSoft, USA) and PAST 3.03 (http://folk.uio.no/ohammer/past/). Differences in site properties were tested by a one-way analysis of variance (ANOVA) followed by the Fisher post hoc test. Because the actinobacterial genome abundance data did not show a normal distribution, the nonparametric Mann-Whitney U test was used to compare the abundances of taxa among sites. A Mantel test with 9999 permutations was used to explore the relationships between community similarity and environmental parameters, either the heavy metal content or the soil Cox, Ntot and pH values. A Bray-Curtis similarity was used as a measure of similarity in the actinobacterial abundance matrix, while Euclidean distances were used for soil chemistry and heavy metal content data. A Pearson r was used as a measure of linear correlation between variables. In all cases, differences of P < 0.05 were regarded as statistically significant.
Results
In this study, we analysed actinobacterial community composition in five soil samples from heavy metal-contaminated sites near a polymetallic smelter. The sites were located along a gradient of contamination by multiple heavy metals, most importantly, Cd, Cu, Zn and Pb. The concentrations of these metals at site S1, located 2500 m from the smelter, were 1.2 ± 0.1, 9.3 ± 0.1, 84.1 ± 3.4 and 160 ± 2 mg kg−1, respectively, and represented background values for the larger area of several tens of km around the smelter. At the most polluted site S5, located 300 m from the metal smelter, the concentrations of the above heavy metals were 8.3 ± 0.1, 22.0 ± 1.0, 272 ± 15 and 1714 ± 62 mg kg−1, respectively, i.e. 7-, 2.3-, 3.2- and 11-fold increases compared with S1 (Table 1). The other environmental properties were rather similar across the sites: pH values ranged from 5.9 to 6.1, Ntot from 0.23 to 0.43 % and Cox from 2.42 to 3.99 %. Soil moisture content measured at the times of sampling ranged between 18 and 32 % and exhibited high variation both within sites and on a seasonal basis. The background site S1 and the most contaminated site S5 did not show significant differences in any of these environmental variables (Table 1).
Quantitative PCR demonstrated that bacterial rRNA gene copy numbers ranged from 0.4 to 3.8 × 109 per gram of soil, and actinobacterial counts were between 0.9 and 8.2 × 108. The lowest mean values were recorded at the two most contaminated sites, S4 and S5, but due to the high variation in the rRNA gene copy numbers within sites, the differences in bacterial and actinobacterial biomasses were not significant among most pairs of sites (Table 1).
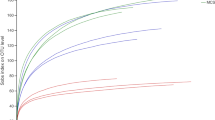
To specifically amplify partial 16S sequences of Actinobacteria, nested PCR was performed using the Actinobacteria-specific primer 16Sact1114r. The in silico test preceding the amplification showed that primer 16Sact1114r was highly specific to Actinobacteria, as in total only 1.8 % of matches came from bacterial genera belonging to other phyla when mismatches were not allowed, 3.1 % in the case of one mismatch, 4.7 % for two mismatches and 10.7 % when three mismatches were allowed. The bacterial phyla with the most false positive hits with up to three mismatches were Proteobacteria (8.3 %; 83 sequences), Firmicutes (18.7 %; 72 sequences) and Cyanobacteria (14 %; 19 sequences). This primer also had high coverage, as it amplified over 97.7 % of the actinobacterial genera present in the dataset with three mismatches allowed, 92.1 % for two mismatches, 90.4 % for three mismatches and 85.1 % when no mismatch was allowed (Supplementary Fig. 1).
The high specificity of primer 16Sact1114r towards Actinobacteria was subsequently confirmed by the results of 454 pyrosequencing, where 16Sact1114r was used in combination with the universal bacterial primer eub530f. In total, 36,381 sequences were obtained by 454 pyrosequencing after the removal of low-quality sequences shorter than 380 bases and potentially chimeric sequences. The clustering of filtered sequences at a 97 % similarity threshold yielded 1305 bacterial OTUs (778 singletons, 59.6 % of all OTUs), where the majority were represented by 1155 actinobacterial OTUs (695 singletons, 60.2 %) that gave the best hits in 147 genera. Only 433 sequences (1.7 % of the total) were identified to belong to other bacterial phyla, mostly to Proteobacteria (55 OTUs) and Firmicutes (52 OTUs). An overview of the taxonomy of the sequences and the OTU richness and abundance at the genus level is shown in Fig. 1.

Overview of abundances and OTU richness of microbial genera from grassland soils across a gradient of heavy metal concentrations obtained by 454 pyrosequencing. Number of OTUs per genus is depicted in greyscale in the inner circle, and a log scale of the relative abundance of genomes per genus is shown by black bars
The analysis of the diversity of soil Actinobacteria showed that even at a relatively shallow depth of 400 sequences per sample, a substantial portion of the diversity was recovered: while the Chao-1 estimates of total OTU richness ranged between 130 and 160, the observed species richness was 85–98 so that between 60 and 70 % of expected diversity was recovered (Table 1). Interestingly, neither the diversity nor the evenness of the actinobacterial community differed significantly between the control plot S1 and the most polluted plot S5 (Table 1). In contrast to the predicted decrease of diversity with the increasing content of heavy metals, the Shannon-Wiener diversity index showed a positive correlation with the concentrations of Cu (P < 0.01), Cd (P < 0.05) and Pb (P < 0.05).
In the most abundant actinobacterial OTUs representing at least 1 % of the estimated number of genomes within a sample for at least three samples, the following genera dominated: Ilumatobacter, Nocardioides, Mycobacterium, Aciditerrimonas, Microlunatus, Actinoallomurus and Pseudonocardia. Genus Ilumatobacter and Aciditerrimonas were also represented by the largest number of OTUs within the whole dataset. These most abundant actinobacterial OTUs and their closest related species are listed in Supplementary Table 1.
The quantitative analysis of actinobacterial and bacterial relative abundance in samples based on qPCR showed that the amount of bacteria negatively correlated with the increasing Cu concentration (P < 0.01) and Zn concentration (P < 0.05), but there was no significant correlation for abundance of Actinobacteria. The Actinobacteria-bacteria ratio appeared to be rising from sites with low contamination to sites with high contamination, but there was high variation in the samples from site S2.
Principal component analysis showed that heavy metals had a slight effect on shaping the actinobacterial community composition (Fig. 2). Despite Pb having the highest concentrations of all of the measured metals at all sites, the most significant effect on the actinobacterial community composition appeared to have been the Cu concentration, as revealed by its strong correlation to most abundant OTUs (Fig. 3). Furthermore, a Mantel test with 9999 permutations indicated a clear relationship between community similarity and heavy metal content (R = 0.3409, P = 0.0001), indicating that actinobacterial community similarity decreased with the increasing difference in heavy metal contents among the samples. The most significant relationships were found between community composition and the content of Cu and Pb, while Zn and Cd showed less of an effect on the actinobacterial community composition. No significant relationship was observed when the actinobacterial community composition was tested against basic soil chemistry data (Cox, Ntot, pH; R = −0.05596, P = 0.8035) or any combination thereof. Among the most abundant actinobacterial taxa, OTUs identified as the members of the genera Acidothermus, Streptomyces, Pseudonocardia, Janibacter and Microlunatus increased in abundance with increasing metal content, while the members of Jatrophihabitans and Actinoallomurus decreased. The genus Ilumatobacter contained several OTUs that responded to heavy metals both positively and negatively (Fig. 3, Supplementary Table 1).

Principal component analysis of relative abundances of genomes of those actinobacterial OTUs representing at least 1 % of total abundance within a sample for at least three samples. Heavy metal concentrations, environmental variables and relative bacterial and actinobacterial abundance are plotted as additional variables

Phylogenetic tree of all actinobacterial OTUs from grassland soils across a gradient of heavy metal concentrations, representing at least 1 % of genome abundance within a sample for at least three samples and their closest identified hits. Relative abundances within the sampling sites are shown by filled circles. Statistically significant differences in correlation between abundance and heavy metal concentration are indicated by +++ for P < 0.001, ++ for P < 0.01 and + for P < 0.1 for positive correlation and similar for negative correlation. (Tree was computed in MEGA6 using Maximum Likelihood method using the parameters as follows. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbour-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among the sites (5 categories (+G, parameter = 0.5976)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 50.3 % sites). The analysis involved 106 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 349 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Tamura et al. 2013).)
Discussion
Because heavy metals constitute the most widespread pollutants in a wide range of environmental contexts, including industrial areas and their surroundings, and because of their cumulative toxicity effects (Brumelis et al. 2002; Hernandez et al. 2003), an investigation of the response of microorganisms to their presence in the environment is important for understanding the potential effects of metal contamination on ecosystem functions. High concentrations of heavy metals are known to negatively affect enzyme activity and are toxic to most microorganisms (Giller et al. 1998). However, there is some evidence that various metals influence the composition of bacterial communities to various extents. Exposure of soil bacterial communities to Cr and As ions significantly lowered their diversity and species richness and changed their community composition (Sheik et al. 2012), while Cd, Pb and especially Zn reduced OTU diversity in polluted soils (Golebiewski et al. 2014; Hur et al. 2011; Moffett et al. 2003). In contrast, long-term Cu exposure was shown to change the bacterial community composition, but not its diversity or species richness (Berg et al. 2012). The results of other studies remain inconclusive because the effects of heavy metals are difficult to separate from the effects of other environmental factors (Chodak et al. 2013).
This study considered soils with a range of heavy metal concentrations which were previously demonstrated to affect soil functions, namely respiration and dehydrogenase activity, which both significantly decreased with the increasing total amount of heavy metals or their extractable fraction (Mühlbachová et al. 2015). We have now demonstrated that the abundance of bacteria estimated using qPCR decreased significantly with the increasing concentrations of Cu and Zn in the soil, while the abundance of Actinobacteria was not affected and the Actinobacteria/bacteria ratio tended to increase (Supplementary Table 2). This result may indicate a lower sensitivity of the Actinobacteria towards metal content. Previous studies on the response of Actinobacteria towards heavy metals remain controversial: despite the fact that some studies using the analyses of phospholipid fatty acid signatures to estimate actinobacterial abundance in soils tended to show that they are less resistant to heavy metals than other bacteria or fungi (Kelly et al. 2003; Zelles 1997), whereas sequencing studies strongly suggest the opposite. The relative abundance of Actinobacteria-dominating soils across the Cu gradient remained stable with increasing bioavailable Cu (Berg et al. 2012), and the sites with increasing heavy metal content that were revegetated using Paulownia fortune showed a higher richness and diversity of Actinobacteria (Liu et al. 2013). Gremion et al. (2003) have shown evidence that the actinomycetes species dominate over all prokaryote diversity in heavy metal-contaminated soil by a combined analysis of 16S rDNA and 16S rRNA, indicating that they might be a dominant part of the metabolically active bacteria in heavy metal-contaminated soils.
In the light of the uncertainty of the response of Actinobacteria to metal toxicity, the actinobacterial communities are an interesting target for exploration. Furthermore, the analysis of the response of individual taxa of the Actinobacteria, which had not been performed to date, should also be able to identify whether the response of Actinobacteria is a phylum-specific trait or whether there are differences on the level of individual taxa. To address this task, we have tested the use of Actinobacteria-specific primers to explore their community composition using 454 pyrosequencing at a high resolution. In agreement with the in silico analyses, the amplicon pool obtained with the 530 F/16Sact1114r primers was greatly dominated by actinobacterial sequences that represented over 98 % of the total. Even with a relatively shallow sampling, more than 60 % of the total estimated actinobacterial diversity was recovered from the study sites.
The results of this study showed that the diversity of Actinobacteria was unaffected by heavy metal content, and a similar diversity was recorded in control plots as well as in those with the highest heavy metal content (Table 1). This finding is in contrast to the theoretical expectation that only a limited number of taxa would survive at highly contaminated sites. Furthermore, the results indicated that major actinobacterial taxa are rarely missing from the highly contaminated sites, although their abundance was affected (Supplementary Table 1).
Despite the fact that total diversity of Actinobacteria was not affected by metal contamination in the studied grassland soils, heavy metals represented the most important factor in shaping their community composition. Although Pb content reached the highest values in the studied sites, it was Cu that most affected the community composition. The abundance of several genera positively correlated with its concentration, especially members of Acidothermus, Streptomyces, Pseudonocardia, Janibacter and Microlunatus. Members of the genera Streptomyces and Janibacter were previously studied for their Cu and Sb resistance (Shi et al. 2013), and the heavy metal resistance of certain Streptomyces spp. is known (Schmidt et al. 2005). To survive in contaminated areas, Streptomyces use heavy metal-protective mechanisms such as a lead-absorbing superoxide dismutase described in Streptomyces subrutilus, allowing them to carry up to 1100 lead atoms per subunit, thus providing highly effective protection against Pb toxicity (So et al. 2001). Probably other members of this genus use similar mechanisms to survive in Pb-contaminated soils, notably because lead is often the main contaminant in mining and smelting areas, as in the case of this study. There are also multiple reports on their ability to degrade lignocellulose (Crawford 1978; Chater et al. 2010; Petrosyan et al. 2002; Větrovský et al. 2014), and together with members from genus Acidothermus, which also includes efficient degraders of cellulose, e.g. Acidothermus cellulolyticus (Blumer-Schuette et al. 2014; Parales et al. 2014) and Microlunatus, which are also involved in the decomposition of plant biomass (Fan et al. 2014), these taxa could be potentially responsible for important organic matter degrading processes in heavy metal-contaminated soils. In addition, the abundance of Pseudonocardia was high in the study area and positively correlated with the heavy metal content. The members of this genus are known as degraders of various organic compounds; Pseudonocardia dioxanivorans is able to use 1,4-dioxane as a sole carbon source, and its growth is stimulated by the addition of Zn (Pornwongthong et al. 2014). In contrast, Jatrophihabitans and Actinoallomurus represented the genera with a significant negative response to heavy metal content. Members of both genera were described as endophytes of plants, which were isolated mainly from plant roots (Madhaiyan et al. 2013; Matsumoto et al. 2012), and thus, the effects of metals on their occurrence might potentially be mediated by the effects on their plant hosts.
This study demonstrated the potential of specific amplification combined with next-generation sequencing to analyse the actinobacterial community composition and its response to mixed metal contamination. The actinobacterial community composition appeared not to be affected by metals in terms of diversity, but the contamination significantly affected community composition. Still, Actinobacteria seem to be less affected by heavy metals than other bacteria, and considering their metabolic potential, they can provide important soil functions, such as a contribution to organic matter decomposition. In this respect, these heavy metal-resistant taxa represent an interesting target for selective exploration.
References
Abdulla HM, El-Shatoury SA (2007) Actinomycetes in rice straw decomposition. Waste Manag 27:850–853
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in-situ detection of individual microbial-cells without cultivation. Microbiol Rev 59:143–169
Anderson I, Abt B, Lykidis A, Klenk HP, Kyrpides N, Ivanova N (2012) Genomics of aerobic cellulose utilization systems in Actinobacteria. PLoS One 7, e39331
Bachar A, Al-Ashhab A, Soares MIM, Sklarz MY, Angel R, Ungar ED, Gillor O (2010) Soil microbial abundance and diversity along a low precipitation gradient. Microb Ecol 60:453–461
Bajkic S, Narancic T, Dokic L, Dordevic D, Nikodinovic-Runic J, Moric I, Vasiljevic B (2013) Microbial diversity and isolation of multiple metal-tolerant bacteria from surface and underground pits within the copper mining and smelting complex bor. Arch Biol Sci 65:375–386
Baldrian P, Kolarik M, Štursová M, Kopecký J, Valášková V, Větrovský T, Žifčáková L, Šnajdr J, Rídl J, Vlček Č, Voříšková J (2012) Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6:248–258
Berg J, Brandt KK, Al-Soud WA, Holm PE, Hansen LH, Sorensen SJ, Nybroe O (2012) Selection for Cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term Cu exposure. Appl Environ Microbiol 78:7438–7446
Berlemont R, Martiny AC (2013) Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microbiol 79:1545–1554
Blumer-Schuette SE, Brown SD, Sander KB, Bayer EA, Kataeva I, Zurawski JV, Conway JM, Adams MWW, Kelly RM (2014) Thermophilic lignocellulose deconstruction. FEMS Microbiol Rev 38:393–448
Brumelis G, Lapina L, Nikodemus O, Tabors G (2002) Use of the O horizon of forest soils in monitoring metal deposition in Latvia. Water Air Soil Pollut 135:291–309
Chater KF, Biro S, Lee KJ, Palmer T, Schrempf H (2010) The complex extracellular biology of Streptomyces. FEMS Microbiol Rev 34:171–198
Chodak M, Golebiewski M, Morawska-Ploskonka J, Kuduk K, Niklinska M (2013) Diversity of microorganisms from forest soils differently polluted with heavy metals. Appl Soil Ecol 64:7–14
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2005) The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33:D294–D296
Crawford DL (1978) Lignocellulose decomposition by selected streptomyces strains. Appl Environ Microbiol 35:1041–1045
de Boer W, Folman LB, Summerbell RC, Boddy L (2005) Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29:795–811
Dominguez-Mendoza CA, Bello-Lopez JM, Navarro-Noya YE, de Leon-Lorenzana AS, Delgado-Balbuena L, Gomez-Acata S, Ruiz-Valdiviezo VM, Ramirez-Villanueva DA, Luna-Guido M, Dendooven L (2014) Bacterial community structure in fumigated soil. Soil Biol Biochem 73:122–129
Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS (2008) Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8:125
Dunbar J, Takala S, Barns SM, Davis JA, Kuske CR (1999) Levels of bacterial community diversity in four arid soils compared by cultivation and 16S rRNA gene cloning. Appl Environ Microbiol 65:1662–1669
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Enkhbaatar B, Temuujin U, Lim JH, Chi WJ, Chang YK, Hong SK (2012) Identification and characterization of a xyloglucan-specific family 74 glycosyl hydrolase from Streptomyces coelicolor A3(2). Appl Environ Microbiol 78:607–611
Fan FL, Yin C, Tang YJ, Li ZJ, Song A, Wakelin SA, Zou J, Liang YC (2014) Probing potential microbial coupling of carbon and nitrogen cycling during decomposition of maize residue by C-13-DNA-SIP. Soil Biol Biochem 70:12–21
Giller KE, Witter E, McGrath SP (1998) Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol Biochem 30:1389–1414
Golebiewski M, Deja-Sikora E, Cichosz M, Tretyn A, Wrobel B (2014) 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microb Ecol 67:635–647
Gremion F, Chatzinotas A, Harms H (2003) Comparative 16S rDNA and 16S rRNA sequence analysis indicates that Actinobacteria might be a dominant part of the metabolically active bacteria in heavy metal-contaminated bulk and rhizosphere soil. Environ Microbiol 5:896–907
Hernandez L, Probst A, Probst JL, Ulrich E (2003) Heavy metal distribution in some French forest soils: evidence for atmospheric contamination. Sci Total Environ 312:195–219
Hur M, Kim Y, Song HR, Kim JM, Choi YI, Yi H (2011) Effect of genetically modified poplars on soil microbial communities during the phytoremediation of waste mine tailings. Appl Environ Microbiol 77:7611–7619
Ivshina IB, Kuyukina MS, Kostina LV (2013) Adaptive mechanisms of nonspecific resistance to heavy metal ions in alkanotrophic actinobacteria. Russ J Ecol 44:123–130
Kalac P, Burda J, Staskova I (1991) Concentrations of lead, cadmium, mercury and copper in mushrooms in the vicinity of lead smelter. Sci Total Environ 105:109–119
Katoh K, Asimenos G, Toh H (2009) Multiple alignment of DNA sequences with MAFFT. In: Posada D (ed) Bioinformatics for DNA sequence analysis, vol 537. Methods Mol Biol. p 39–64
Kelly JJ, Haggblom MM, Tate RL (2003) Effects of heavy metal contamination and remediation on soil microbial communities in the vicinity of a zinc smelter as indicated by analysis of microbial community phospholipid fatty acid profiles. Biol Fertil Soils 38:65–71
Kyselkova M, Kopecky J, Felfoldi T, Cermak L, Omelka M, Grundmann GL, Moenne-Loccoz Y, Sagova-Mareckova M (2008) Development of a 16S rRNA gene-based prototype microarray for the detection of selected actinomycetes genera Antonie Van Leeuwenhoek. Int J Gen Mol Microbiol 94:439–453
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120
Leigh MB, Pellizari VH, Uhlik O, Sutka R, Rodrigues J, Ostrom NE, Zhou JH, Tiedje JM (2007) Biphenyl-utilizing bacteria and their functional genes in a pine root zone contaminated with polychlorinated biphenyls (PCBs). ISME J 1:134–148
Liu W, Wang J, Zhang CB (2013) Evaluation of soil chemical properties and actinomycete community structure following a temporal sequence of revegetation through Paulownia fortunei in the heavy metal-contaminated soil. Water Air Soil Pollut 224:1730
Madhaiyan M, Hu CJ, Kim SJ, Weon HY, Kwon SW, Ji LH (2013) Jatrophihabitans endophyticus gen. nov., sp nov., an endophytic actinobacterium isolated from a surface-sterilized stem of Jatropha curcas L. Int J Syst Evol Microbiol 63:1241–1248
Matsumoto A, Fukuda A, Inahashi Y, Omura S, Takahashi Y (2012) Actinoallomurus radicium sp. nov., isolated from the roots of two plant species. Int J Syst Evol Microbiol 62:295–298
Moffett BF, Nicholson FA, Uwakwe NC, Chambers BJ, Harris JA, Hill TCJ (2003) Zinc contamination decreases the bacterial diversity of agricultural soil. FEMS Microbiol Ecol 43:13–19
Mühlbachová G (2011) Soil microbial activities and heavy metal mobility in long-term contaminated soils after addition of EDTA and EDDS. Ecol Eng 37:1064–1071
Mühlbachová G, Ságová-Marečková M, Omelka M, Szákova J, Tlustoš P (2015) The influence of soil organic carbon on interactions between microbial parameters and metal concentrations at a long-term contaminated site. Sci Total Environ 502:218–223
Muyzer G, Dewaal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Parales RE, Berry AM, Parales JV, Barabote RD (2014) Use of acidothermus cellulolyticus xylanase for hydrolyzing lignocellulose. US Patent No 08778649 B2
Petrosyan P, Luz-Madrigal A, Huitron C, Flores ME (2002) Characterization of a xylanolytic complex from Streptomyces sp. Biotechnol Lett 24:1473–1476
Pornwongthong P, Mulchandani A, Gedalanga PB, Mahendra S (2014) Transition metals and organic ligands influence biodegradation of 1,4-dioxane. Appl Biochem Biotechnol 173:291–306
Sagova-Mareckova M, Cermak L, Novotna J, Plhackova K, Forstova J, Kopecky J (2008) Innovative methods for soil DNA purification tested in soils with widely differing characteristics. Appl Environ Microbiol 74:2902–2907
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Schmidt A, Haferburg G, Sineriz M, Merten D, Buchel G, Kothe E (2005) Heavy metal resistance mechanisms in actinobacteria for survival in AMD contaminated soils. Chem Erde-Geochem 65:131–144
Sheik CS, Mitchell TW, Rizvi FZ, Rehman Y, Faisal M, Hasnain S, McInerney MJ, Krumholz LR (2012) Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS One 7, e40059
Shi ZJ, Cao Z, Qin D, Zhu WT, Wang Q, Li MS, Wang GJ (2013) Correlation models between environmental factors and bacterial resistance to antimony and copper. PLoS One 8:78533
So NW, Rho JY, Lee SY, Hancock IC, Kim JH (2001) A lead-absorbing protein with superoxide dismutase activity from Streptomyces subrutilus. FEMS Microbiol Lett 194:93–98
Stach JEM, Maldonado LA, Ward AC, Goodfellow M, Bull AT (2003) New primers for the class Actinobacteria: application to marine and terrestrial environments. Environ Microbiol 5:828–841
Steger K, Sjogren AM, Jarvis A, Jansson JK, Sundh I (2007) Development of compost maturity and Actinobacteria populations during full-scale composting of organic household waste. J Appl Microbiol 103:487–498
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30:2725–2729
Větrovský T, Baldrian P (2013a) Analysis of soil fungal communities by amplicon pyrosequencing: current approaches to data analysis and the introduction of the pipeline SEED. Biol Fertil Soils 49:1027–1037
Větrovský T, Baldrian P (2013b) The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:57923
Větrovský T, Steffen KT, Baldrian P (2014) Potential of cometabolic transformation of polysaccharides and lignin in lignocellulose by soil Actinobacteria. PLoS One 9:89108
Wang YX, Liu Q, Yan L, Gao YM, Wang YJ, Wang WD (2013) A novel lignin degradation bacterial consortium for efficient pulping. Bioresour Technol 139:113–119
Wilmotte A, Vanderauwera G, Dewachter R (1993) Structure of the 16 S ribosomal RNA of the thermophilic cyanobacterium Chlorogloeopsis HTF (‘Mastigocladus laminosus HTF’) strain PCC7518, and phylogenetic analysis. FEBS Lett 317:96–100
Yin LJ, Huang PS, Lin HH (2010) Isolation of cellulase-producing bacteria and characterization of the cellulase from the isolated bacterium Cellulomonas Sp YJ5. J Agric Food Chem 58:9833–9837
Zelles L (1997) Phospholipid fatty acid profiles in selected members of soil microbial communities. Chemosphere 35:275–294
Acknowledgments
This work was supported by the Academy of Sciences of the Czech Republic (IAA603020901), by the Ministry of Education, Youth and Sports of the Czech Republic (LD12048, LD12050) and by the research concept of the Institute of Microbiology of the ASCR, v.v.i. (RVO61388971). This publication was also supported by the project ‘BIOCEV–Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University’
(CZ.1.05/1.1.00/02.0109) from the European Regional Development Fund. The authors are thankful to the anonymous reviewers and the editor of the manuscript whose comments helped to improve the content of the paper.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
(PDF 73 kb)
Supplementary Table 1
(PDF 109 kb)
Supplementary Table 2
(PDF 51 kb)
Rights and permissions
About this article

Cite this article
Větrovský, T., Baldrian, P. An in-depth analysis of actinobacterial communities shows their high diversity in grassland soils along a gradient of mixed heavy metal contamination. Biol Fertil Soils 51, 827–837 (2015). https://doi.org/10.1007/s00374-015-1029-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-015-1029-9