
Abstract
To understand the organic sulfur (S) stabilization in volcanic soils, we investigated organic S transformation rates and their relationships to soil properties in incubation experiments using forest soils from the Nikko volcanic region, central Japan. We hypothesized that carbon (C)-bonded S would first be transformed into ester sulfate-S and then into inorganic sulfate-S. We separately calculated the rates of decrease of C-bonded S (velocity 1, v 1) and ester sulfate-S (velocity 2, v 2) concentrations. During incubation, the ester sulfate-S concentration increased in two soils characterized by a high concentration of both ammonium oxalate-extractable aluminum (Alo) and pyrophosphate-extractable Al (Alp), whereas the C-bonded S concentration decreased in all soils. A large proportion of the S that was lost in the incubation experiments consisted of C-bonded S rather than ester sulfate-S. Velocity 2 was negatively correlated with both of Alo and Alp contents when soils were incubated at 20 °C. These results suggest that when C-bonded S is transformed into ester sulfate-S, complete mineralization to inorganic sulfate is inhibited, because ester sulfate-S is stabilized due to organo–mineral association. Incubation temperatures significantly affected v 2. Thus, production of inorganic sulfate by mineralization of ester sulfate-S appeared to be regulated by soil Al contents and temperatures. Velocity 1 was proportional to soil pH ranging from 4.5 to 5.5, indicating that the degradation of C-bonded S is pH dependent.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfur, an essential element for plants and microorganisms, is continuously cycled between organic and inorganic forms (Castellano and Dick 1990). Mineralization of organic S is a key process for regulating the S cycling in forest soils. Traditionally, soil organic S has been fractionated into hydriodic acid (HI)-reducible and non-reducible S, which are thought to represent ester sulfates and C-bonded S, respectively (Freney 1986). The former contains the C–O–S bonds (ester sulfate) and the C–N–S bonds (sulfamate) along with some organic sulfites (Freney 1961) and corresponds to highly oxidized soil organic S (Solomon et al. 2011). The latter contain organic S directly bonded to C (C–S) and correspond to highly reduced S (e.g., organic sulfides and thiols) and S with intermediate oxidation states (e.g., sulfonates and sulfoxides) (Solomon et al. 2011). Mineralization process of C-bonded S is different from that of ester sulfates (McGill and Cole 1981), and it is a two-step process in which C-bonded S is first converted into ester sulfates, followed by hydrolysis of the ester sulfates (McGill and Cole 1981; Ghani et al. 1991, 1992; Norman et al. 2002; Solomon et al. 2003, 2011; Churka Blum et al. 2013). Nevertheless, the sequential transformation rates, i.e., “change rates of S from C-bonded S to ester sulfate-S” and “change rates from ester sulfate-S to inorganic sulfate-S,” have not been separately estimated yet; furthermore, influence of soil properties on mineralization of C-bonded S and ester sulfate-S are poorly known. The occurrence of the ester sulfate-S mineralization can be obscured by the C-bonded S mineralization to sulfate and subsequent transformation to ester sulfate-S (Ghani et al. 1991). If the sequential transformation rates are estimated separately, the mineralization rate of ester sulfate-S is deleted from S mineralized from C-bonded S form and this can allow studying effects of climate and soil properties on both S mineralization rates.
The ester sulfate-S fraction is believed to consist primarily of organic compounds with widely different solubility, ranging from protected clay-associated forms to highly soluble ester sulfates (Eriksen et al. 1998). Clays can physically and chemically stabilize organic S directly or indirectly due to strong association of organic C with aluminum (Al) and iron (Fe) oxides and hydroxides covering clays. The direct interaction depends on specific charge characteristics and surface area of clays (Solomon et al. 2003). Particularly volcanic soils have surface-active (highly reactive) Al/Fe solid compounds (Yagasaki et al. 2006). We previously observed that Japanese forest soils derived from volcanic materials contained large amount of HI-reducible S, acid ammonium oxalate-extractable S (Soxa), and pyrophosphate-soluble S (Spyp) (Tanikawa et al. 2009a, 2013). Each fraction accounted for more than 60 % of total S concentration, and they were significantly (p < 0.001) correlated with one another, and thus overlapped. Because HI-reducible S comprise of ester sulfate-S and inorganic sulfate-S, the predominant organic S form in both Soxa and Spyp was postulated to be ester sulfate-S. Organically bound S extracted with pyrophosphate was significantly correlated with oxalate-extractable Al (Alo) concentration. The oxalate-extractable Al/Fe act as aggregating agents (Igwe et al. 2009), and pyrophosphate reagent can dissolve Al/Fe–humus complex (McKeague 1967; Wada 1989) with a little portion of the other active Al/Fe forms (Kaiser and Zech 1996; García-Rodeja et al. 2004). Based on the previous studies, the occlusion of ester sulfate-S in soil aggregate and the formation of complexes between ester sulfate-S and metal–humus are probably high in soils containing high amounts of active Al/Fe during organic S transformation.
The aim of the present study was (1) to separately estimate the transformation rates of organic S from C-bonded S into ester sulfate-S (v 1) and of ester sulfate-S into inorganic sulfate-S (v 2), and (2) to evaluate the influence both of site-specific soil properties and incubation temperatures on the transformation rates (v 1 and v 2). It was hypothesized that ester sulfate-S is derived from C-bonded, and then the inhibition of ester sulfate-S mineralization is observed at soil with high contents of active Al. Since stable soil organic matter (SOM) is characterized by low activation energies in the Arrhenius function (Lützow and Kögel-Knabner 2009), v 2 is hypnotized to be more temperature sensitive than v 1. Japanese subalpine soils tend to accumulate high amounts of decomposable SOM, because of their cool temperatures. We predict that these soils contain a high amount of substrates (organic S), and that both long incubation periods and a high temperature can increase the mineralization of organic S as described by Ghani et al. (1991).
The mineralization of organic S is very low at 10 °C but increases by increasing temperature from 20 to 40 °C and then it decreases (Stevenson and Cole 1999). Sulfite oxidase activities have an optimal pH of 8.0 and increase by increasing temperature up to 35 °C (Bilen and Dick 2011). Arylsulfatase activities that hydrolyze of ester sulfate showed an optimal pH at about 6.2, and increase by increasing the temperature up to about 60 °C, with inactivation temperatures ranging from 60 to 70 °C (Tabatabai 1996). Considering all these reports, we chose 20 and 35 °C temperatures sufficiently high for mineralization of ester sulfate-S. The discrete temperature step was chosen to evaluate the temperature dependency of the sequential transformation rates: v 1 and v 2. To investigate the influence of site-specific properties on S transformations, three soils with different chemical properties were selected to have a variation in soil pH and in Al–humus complex concentrations according to our previous study (Tanikawa et al. 2009b).
Materials and methods
Soils
Soils were sampled from subalpine forest sites in the Nikko volcanic region of Tochigi Prefecture, central Japan and the relative chemical properties are reported by Tanikawa et al. (2009b). There are no active fumaroles in this region. We studied soils from three sites, designated as DC, HL, and NA (Table 1). The dominant vegetation at the DC site was bamboo shrubs (Sasa nipponica), which has recently replaced the original coniferous forest of Tsuga diversifolia. The dominant vegetation at the NA site was T. diversifolia, whereas T. diversifolia and Betula ermanii were dominant at the HL site. The soils were classified as Andic Dystrudepts according to the USDA Soil Taxonomy (Soil Survey Staff 2010). Three soil samples were randomly collected at each site with an approximate distance of 10 m among them. Each sample consisted of four pooled subsamples. Subsamples were taken from the A1 horizon by a trowel and had a size of 50 cm × 50 cm. The litter layer was removed before soil sampling. All coarse plant materials were removed, and then subsamples were thoroughly mixed to give the composite sample. Then, field-moist soils were sieved (2 mm) and stored in polyethylene bags at 4 °C until they were used in the incubation experiment.
Fresh soil was analyzed for soil pH (H2O) and the phosphate-extractable sulfate-S concentration (Tabatabai 1996), oven-dried to calculate water content, or air-dried before being ground, sieved (149-μm) and analyzed for the initial total S, initial HI-reducible S (which includes the ester sulfate-S and the inorganic sulfate-S), and the organic C and N concentrations.
Microbial incubation experiments
The incubation experiments were performed according to Tabatabai and Al-Khafaji (1980). We placed a layer of fine glass beads (each 350 to 710 μm in diameter) and a thin glass wool pad (about 5 mm thick) in the bottom of a 100-mL plastic syringe (JS-S00S, JMS Co. Ltd., Tokyo, Japan). A field-moist soil sample equivalent to 20 g dry weight was thoroughly mixed with 20 g of coarse glass beads (2.0 to 2.8 mm in diameter) in order to avoid compaction of soils during leaching, maintain aeration, and enhance leaching (Ghani et al. 1991). The mixture was transferred into the syringe. Another glass wool pad was used to prevent dispersal of soil when the KCl solution (described below) was poured over the soil–glass beads column. A silicon tube was attached to the lower end of the syringe for collecting the leachate. When the incubation experiment was started, the soil–glass beads mixture was leached with 100 ml 0.01 M KCl in four to five times to remove inorganic sulfate-S from soil, and excess water was removed under vacuum at 67 kPa. The concentration of the KCl solution was chosen considering that microbial (e.g., fungi) biomass and growth rates did not significantly decrease at salt concentrations lower than 0.2 M (e.g., Matsuda et al. 2006). The top of the syringe was then covered with a piece of plastic film (Saran Wrap) having several holes created with a wooden pick to provide aeration; the syringe was set in an upright position in a rack.
We prepared a total of nine syringes (three soil samples at each the site) for incubation at both 20 and 35 °C. The incubation lasted for 2 weeks from the initial preparation and leaching of the syringes; then the soil mixture was leached again with 0.01 M KCl solution, the plastic film was removed, and suction was applied to produce a vacuum of 67 kPa. The syringe was again covered with the plastic film, placed vertically in the rack, and incubated at the chosen temperature. The leaching procedure was repeated every 2-week intervals and the incubation lasted for 40 weeks. The water contents of soils in the incubation syringes were adjusted by weighing them every week and adding deionized water to maintain a constant weight. At the end of the incubation experiment, a portion of each subsample incubated was oven-dried to calculate the water content.
Before incubation, the gravimetric soil water content was 58, 53, and 57 % in the DC, HL, and NA soils, respectively. After the first leaching and the removal of excess water by vacuum, the water content increased to 67, 64, and 66 % in the DC, HL, and NA soils, respectively. After the second leaching procedure, the water content decreased to levels similar to those before the beginning of incubation: 59 % for the DC soil, 53 % for the HL soil, and 57 % for the NA soil. The stable water contents were then maintained until the end of the incubation period. During the incubation period, the average water contents (n = 3) were 59 ± 2 % for the DC soil, 54 ± 3 % for the HL soil, and 58 ± 2 % for the NA soil at 20 °C and 60 ± 2 % for the DC soil, 55 ± 3 % for the HL soil, and 58 ± 2 % for the NA soil at 35 °C. The soil water content at the end of the incubation was similar to the initial water content; 59, 53, and 58 % in the DC, HL, and NA soils, respectively.
The leachates were filtered (0.45 μm Chromato Disk, 13AI, Kurabo Industries Ltd., Osaka, Japan), then we measured the sulfate-S concentrations by ion chromatography (IC 7000 Series II, Yokogawa Analytical Systems, Tokyo, Japan).
Chemical analysis
Table 2 lists the methods used to determine the soil S fractions. The soil S concentrations were measured as described by Tabatabai (1996). The ground soil sample was oxidized with an HNO3–HClO4 mixture, and then the initial total S concentration in the digested material was determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) (Maxim, Fisons Instruments, Ecublens, Switzerland). The final total S was calculated by subtracting “the cumulative S mass leached from soil” from the initial total S concentration to avoid an overestimation of mineralized S amount due to soluble organic S.
The ester sulfate-S of the ground soil samples was directly reduced with HI (Freney 1961). After reduction to sulfide, the concentration of S was colorimetrically determined by the methylene blue method (Johnson and Nishita 1952). Because the soil solution and the adsorbed sulfate-S are also reduced by HI (Tabatabai 1996), the ester sulfate-S fraction was obtained by subtracting Sphos, determined as described below, from the HI-reducible S.
The C-bonded S concentration was obtained by subtracting the initial ester sulfate-S and the initial Sphos from the initial total S (Freney 1967). Because the adsorbed S and the soil solution sulfate-S are usually extracted with phosphate solution (Kowalenko 1993), we determined Sphos by shaking field-moist soil samples (5 g of oven-dried soil) with 50 ml 500 mg P Ca(H2PO4)2 H2O L–1 for 30 min (Maynard et al. 1987); then the mixture was filtered through no. 6 filter paper (Advantec, Dublin, CA, USA). The concentration of sulfate-S in the extract was determined by ion chromatography (IC 7000 Series II). The pH of the phosphate solvent never exceeded 7, so the extraction of organic S was minimized (Kowalenko 1993).
Sulfate-S is the main inorganic form in most soils, with elemental and sulfide forms present in soils under predominantly anaerobic conditions (Kowalenko 1993). In an analysis of well-drained, well-aerated soils, reduced inorganic S compounds are not expected (Tabatabai 1996). Therefore, we estimated the total concentration of soil inorganic sulfate-S of the soil solution and adsorbed sulfate-S, which are the dominant Sphos fractions. Organic S concentration was calculated by subtracting Sphos concentration from total S concentration.
The total C and N concentrations were measured using a CN analyzer (MT600 CN Corder, Yanaco Ltd., Kyoto, Japan). Because the amount of inorganic C is negligible in noncalcareous soils (Nelson and Sommers 1996), we regarded total C as the organic C fraction of our samples. Soil pH (H2O) was determined with a glass electrode using fresh soil samples (soil:water = 1:2.5 w/w). Extractable Al was determined following the methods of the International Soil Reference and Information Centre (ISRIC 1993). Alo was extracted with 0.2 M acid ammonium oxalate solution by shaking air-dried soil (pH 3; soil:solution = 1:50 w/w) for 4 h in the dark. The Al concentrations in the extracts were determined by ICP-AES after centrifugation (8,000–9,000 × g for 1 h) with three drops of Superfloc and filtration through a 0.45-μm membrane filter. Oxalate extracts Al in hydrous amorphous to paracrystalline aluminosilicate minerals (allophane, allophane-like minerals, and imogolite) and in humus complex. The pyrophosphate-extractable Al (Alp) was extracted with 0.1 M Na4P2O7 10H2O by shaking air-dried soil (soil:solution = 1:100 w/w) for 16 h. Pyrophosphate extraction can be used to extract Al from Al–humus complexes (Higashi and Shinagawa 1981), and strong correlation between Alp and total organic carbon is observed in volcanic soils (e.g., Matus et al. 2008). However, the solvent can extract small amount of aluminous goethite (Andriesse 1979), amorphous Al hydroxides and gibbsite (Kaiser and Zech 1996), some interlayer complexed Al in the silt plus clay, or relatively labile forms of surface-precipitated Al-OM (García-Rodeja et al. 2004). These overestimations of Al–humus complexes might be serious when the suspension is low-speed centrifuged without Superfloc and then the supernatant is not adopted for ultrafiltration (Schuppli et al. 1983). Therefore, the suspensions were centrifuged at high speed for a long time (8,000–9,000 × g for 1 h) with three drops of Superfloc and then passed through a 0.025-μm Millipore filter (Schuppli et al. 1983) in the present study. All values are reported on an oven-dry mass basis.
Calculations and statistical analyses
In this study, we defined words related to “transformation” as follows: “S mineralization” for ultimate formation of inorganic sulfate-S from ester sulfate-S and C-bonded S; “transformation of C-bonded S” for change of C-bonded S into ester sulfate-S; “degradation of C-bonded S” for change of C-bonded S into ester and inorganic sulfate-S; “mineralization of ester sulfate-S” for change of ester sulfate-S into inorganic sulfate-S. “The total mass of sulfate-S produced during the incubation” was calculated as the sum of “the cumulative S mass leached from soil” and “the increment of dissolved and adsorbed sulfate-S in soils after the incubation (ΔSphos, determined as described below).” The overall mineralization rate of organic S (OMR) was obtained by dividing “the total mass of sulfate-S produced during the incubation” by 280 days. The results are reported as the average of three replicates on an oven-dry basis. The leached sulfate-S at the beginning of the incubation period after the first application of the KCl solution accounted for 8 to 29 % of total leaching mass of sulfate-S, and was excluded from “the cumulative S mass leached from soil.” The decrease in the S fractions after incubation (Δtotal S, ΔC-bonded S, and Δester sulfate-S) was calculated by subtracting the final concentration of a given fraction from the initial concentration of that fraction. The increment of Sphos (ΔSphos) was calculated by subtracting the initial Sphos from the final Sphos.
The transformation rate of C-bonded S (v 1) was calculated by the following equation:
where t is time.
The mineralization of ester sulfate-S (v 2) was calculated by the following equation:
By rearranging both equations, v 1 and v 2 can be calculated as:
Immobilization of inorganic sulfate-S and conversion of ester sulfate-S into C-bonded S have been observed in previous studies (e.g., McLaren et al. 1985; Castellano and Dick 1990). Therefore, the transformation rates v 1 and v 2 are net but not gross rates. If the conversion rates from ester sulfate-S and inorganic sulfate-S into C-bonded S are higher than degradation of C-bonded S into ester sulfate-S and inorganic sulfate-S, v 1 would be negative. Similarly, v 1 and v 2 would be negative when net immobilization of inorganic S occurs. Direct mineralization of C-bonded S into inorganic sulfate-S would lead to overestimation of v 1 and subsequent overestimation of v 2. Therefore, v 1 and v 2 likely represent the maximum net rates.
To test whether the concentrations of the initial total S and initial S fractions differed significantly among soils, we used the χ2 test. Paired t test was performed for detecting differences between initial C-bonded S concentrations and initial ester sulfate-S concentrations of soils. We tested for significant differences among sites and incubation temperatures in terms of their effects on OMR, Δester sulfate-S, ΔC-bonded S, v 1, and v 2 using a split-plot ANOVA design, with the site as the main plot and temperature as the subplot. We used regression analysis to determine whether the relationships between v and soil pH, between v and Alp, and between v and Alo were statistically significant. All statistical analyses were performed using version 06 J of the STATISTICA software (StatSoft, Tulsa, OK, USA).
Results
Initial soil S fractions
The initial total S concentration ranged from 820 to 1,270 mg S kg–1. The dominant S fraction was organic S ranging from 803 to 1,190 mg S kg–1. The initial ester sulfate-S concentration ranged from 301 to 769 mg S kg−1, as same as the initial C-bonded S concentration from 426 to 502 mg S kg−1. The initial Sphos was a minor fraction since values ranged from 17 to 79 mg S kg–1. The concentrations of total S, ester sulfate-S, C-bonded S, and Sphos differed significantly among soils (p < 0.05). The initial total S, organic S, ester sulfate-S, and Sphos concentrations of the DC soil were the highest. The C:S ratio ranged from 112 to 157 and Alp was from 9.0 to 11.6 g Al kg–1 (Table 1).
The overall mineralization rates of organic S, v1 and v2
“The cumulative S mass leached from soil” increased with incubation period (Fig. 1) as previously reported (Pirela and Tabatabi 1988; Ghani et al.1991). In our study, OMR ranged from 0.03 to 0.45 mg S kg–1 day–1 and fell within the range of values of previous studies (Tables 3 and 4). Among our soils and data of previous studies (Tables 3 and 4), the DC soil had the lowest OMR value at 20 °C. The v 1 values can be ranked as NA > DC > HL at both temperatures, whereas the rank order of v 2 values was HL > NA > > DC at both temperatures (Table 5). All rates were of the same order of magnitude, except for v 2 at 20 °C in the DC soil, which was an order of magnitude smaller than the other rates. The difference between v 1 and v 2 values was not significant at each temperature.

The cumulative S mass leached from the soil during the incubation experiments (mean ± SE; n = 3). a 20 °C; b 35 °C
Influence of site
While the site influence on OMR was not significant, ΔC-bonded S, Δester sulfate-S, v 1, and v 2 differed significantly among sites (p < 0.05).
The C-bonded S concentration decreased in all soils during incubation (Fig. 2a), whereas the ester sulfate-S concentration increased during incubation in the DC and NA soils (Fig. 2b), which had a high Alp. The ΔC-bonded S concentration ranged from 68 to 139 mg S kg–1 at 20 °C and from 62 to 145 mg S kg–1 at 35 °C. The Δester sulfate-S concentration ranged from −79 to 58 mg S kg–1 at 20 °C and from −9.2 to 82 mg S kg–1 at 35 °C. A large proportion of the S that was lost in the incubation experiments consisted of C-bonded S rather than ester sulfate-S, because the ratio of ΔC-bonded S/Δtotal S was higher (89 to 185 % at 20 °C, 52 to 103 % at 35 °C) at both incubation temperatures than the ratio of Δester sulfate-S/Δtotal S (−151 to 75 % at 20 °C and from −6 to 66 % at 35 °C).

The amounts of the organic S fractions decreased during the incubation experiments (mean ± SE; n = 3). DC, HL, and NA are names of the study sites (see Table 1). a C-bonded S; b ester sulfate-S
Relationships between the sequential transformation rates of organic S and soil properties
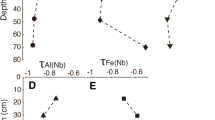
Velocity 1 but not v 2 was directly proportional to soil pH (Fig. 3a, b), as shown by the following regression equations at both incubation temperatures:

Relationships between site-specific properties and v 1 or v 2 (mean ± SE; n = 3). a soil pH (H2O) and v 1; b soil pH (H2O) and v 2; c Alo content and v 1; d Alo content and v 2; e Alp content and v 1; f Alp content and v 2
At 20 °C:
At 35 °C:
Velocity 2 was also significantly correlated with Alo at the 20 °C incubation temperature:
Similarly, a significant regression equation was obtained between v 2 and Alp at the 20 °C incubation temperature:
Influence of temperature
In all three soils, OMR was significantly higher during incubation at 35 °C than at 20 °C (p = 0.002, Table 3). Although Δester sulfate-S concentration differed significantly between the two temperatures (p = 0.012), that of ΔC-bonded S did not (Fig. 2a, b). Likewise, v 1 did not differ significantly between the two temperatures, but v 2 at 20 °C was significantly lower than at 35 °C (p = 0.003) (Table 5).
Discussion
Transformation pathways
Increases of ester sulfate-S concentrations and decreases of C-bonded S concentrations during the incubation in the DC and NA soils suggested that C-bonded S is first transformed to ester sulfate-S before formation of inorganic sulfate-S, as described by McGill and Cole (1981), Ghani et al. (1991, 1992), and Solomon et al. (2003, 2011). The fact that the ratio of ΔC-bonded S/Δtotal S was higher than the ratio of Δester sulfate-S/Δtotal S indicated that the total mass of sulfate-S produced during incubation was derived almost exclusively from C-bonded S, being consistent with earlier studies (e.g., Ghani et al. 1991; Zhao et al. 2006; Solomon et al. 2011). In forests, plants absorb S mostly sulfate-S (Kertesz and Mirleau 2004) and synthesize organic S in the leaves, and the organic S is released into soils via litter and root exudates. Schroth et al. (2007) revealed that most of S present in plant litter is C-bonded S, accounting for more than 80 % of organic S. Roots exude amino acids, aliphatic acids, and proteins (Badri et al. 2009), being C-bonded S the predominant organic S form. During litter decomposition process, oxidation of organic S occurs and enriches this pool with oxidized compounds (Schroth et al. 2007); this process is accelerated by anthropogenic activities such as cultivation (Solomon et al. 2011). Our result also supported that the degradation of soil organic S originated from plants (C-bonded S) is generally assumed to represent a shift from reduced to oxidized forms (ester sulfate-S).
Because v 2 was positive in all soils (Table 5), the immobilization of inorganic sulfate-S into ester sulfate-S was probably a minor pathway. In addition, v 1 was also positive in all soils (Table 5). Therefore, mineralization was superior to immobilization even if immobilization/mineralization turnover was likely rapid as shown by radioisotope 35S experiments (e.g., Eriksen 2005).
Velocity 1 in this study was higher than that calculated from Ghani et al. (1991, 1992) (ranging from 0.1 to 0.3 mg S kg−1 day−1 at 30 °C) who performed a comparable experiment to that of this study using New Zealand soils. The higher rates of v 1 may be explained by the higher amounts of substrates (C-bonded S) in our volcanic soils (ranging from 430 to 500 mg S kg−1) than in New Zealand soils (ranging from 83 to 368 mg S kg−1).
Influence of active Al on transformation rates
To our knowledge, this is the first study to estimate the sequential S transformation rates, v 1 and v 2. By the estimation of v 1 and v 2 separately, significant site influences on both v 1 and v 2 were observed. Velocity 2 was significantly (p < 0.05) and negatively correlated with both of Alo and Alp contents at 20 °C (Fig. 3d, f), suggesting that ester sulfate-S is occluded in soil aggregates and adsorbed onto mineral surface (incorporation into Al–humus complexes). Bettany et al. (1979) suggested that ester sulfate-S is associated with the active side chain components of fulvic and humic materials. Ester-bonded sulfate groups can serve as complexation sites for metals such as Al and Fe (Sollins et al. 1996). Due to these characteristics, ester sulfate-S might be suitable for the occlusion and complexation reactions.
Even though ester sulfate-S is often a minor organic S form in forest soils (e.g., Fitzgerald et al. 1988; Schroth et al. 2007), there was no significant difference between initial ester sulfate-S concentrations and initial C-bonded S concentrations in this study. Furthermore, initial ester sulfate-S concentration in our soils was higher than those reported in bibliography (Mitchell et al. 1992). Because great amounts of Al/Fe are released by the weathering of volcanic ash, a large amount of them is accumulated as hydroxides or metal–humus complexes (Yoshinaga 1986). The accumulation of ester sulfate-S appeared to be a unique characteristic of Japanese volcanic soils due to the high concentration of active Al. Keer et al. (1990) indicated that ester sulfates were mainly present in the high molecular weight compounds, whereas Lou and Warman (1992) stated that the ester sulfate-S associated with the low molecular weight fraction of SOM can be defolded by enzymatic hydrolysis. Estimation of both molecular weight of organic compounds including ester sulfates and transformation rates (v 1 and v 2) in soils with various textures may resolve this contradiction and supply information on various residence times of ester sulfate-S. The information can help to predict both plant available S and watersheds acidification both due to mobilization of soil organic S.
Influence of soil pH on transformation rates
Velocity 1 was positively correlated with soil pH, which ranged from 4.5 to 5.5 (Fig. 3a), suggesting that a decrease of soil pH suppress the degradation of C-bonded S. This result was similar to previous studies for the overall mineralization rate of organic S but not for C-bonded S; OMR was most rapid at pH ranging from 5.5 to about 7.5 (Stevenson and Cole 1999), and was correlated to pH at values lower than pH 7.5 (Williams 1967). Our result might be caused by pH dependency of microbial activity as shown by Haynes (1986), Pietri and Brookes (2008) and by the pH dependency of enzyme activities in soils such as sulfite oxidase activity (Bilen and Dick 2011).
In contrast, v 2 was not related to soil pH (Fig. 3b). Two reasons for the different reactions between C-bonded S and ester sulfate-S to soil pH can be postulated. First, C-bonded S and ester sulfate-S may have heterogeneous quantitative distributions at hierarchical aggregate structures (e.g., Tisdall and Oades 1982; O'Brien and Jastrow 2013). Since plant residues, living roots, and fungal hypha were distributed in the outer part of the hierarchical aggregate structures (non-aggregated material and macroaggregate), a large portion of C-bonded S may be located in the outer part of the structures. In contrast, ester sulfate-S is mainly generated through biochemical process by soil microorganisms, metabolizing organic residues (Solomon et al. 2001), and most of ester sulfate-S is often found in the clay fraction rather than in the silt fraction (e.g., Bettany et al. 1979; Wang et al. 2006; Prietzel et al. 2007). These observations agree with the idea that ester sulfate-S is distributed in the inner part of the microaggregates. Therefore, C-bonded S may be located and biologically transformed into ester sulfate-S in the outer part of macroaggregates. Then, ester sulfate-S produced may reach the inner part of the aggregate structures due to its affinity to minerals, or be absorbed onto the surface of other organic materials, which are associated to minerals, like the onion layer model proposed for SOM stabilization by Sollins et al. (2006). Because microaggregates provide a unique microenvironment that selects specific microbial species in different soils (Mummey et al. 2006), pH dependency of transformation/mineralization processes may be different between C-bonded S and esters sulfate-S. In addition, different kinds of sulfatase, such as aryl-, alkyl-, steroid, gluco-, condro- and mycosulphatase activities can be active in these processes but only arylsulfatase activity has been investigated in soil (Eriksen 2008). Activities of enzymes interacting with mineral fraction and organo–mineral complexes can have different pH profiles compared to non-immobilized enzyme (Rao et al. 2000; Kelleher et al. 2004), and generally enzyme–mineral complexes have a broad pH optimum (Nannipieri et al. 1988). If several enzymes and the mineral complexes with different pH optimums contribute to the mineralization of ester sulfate-S, synergistically, linear relationships between soil pH and ester sulfate-S mineralization rate cannot be obtained.
Influence of temperature on transformation rates
Temperature is one of the most important factors governing the turnover of soil organic S pool (Wang et al. 2006). In the present study, v 2 is hypnotized to be more temperature sensitive than v 1. This hypothesis was consistent with observations that the temperature significantly affected v 2 and that the relationship between v 2 and Alp was significant at 20 °C but not at 35 °C, suggesting that the ester sulfate-S stabilization of soils is greater at lower than at higher temperatures. Rasmussen et al. (2006) also indicated that the negative correlation between Al hydroxide (including Al–humus complexes) content and C mineralization increased as temperature decreased, suggesting that Al species were important in controlling C mineralization at low temperature. Probably this depended on the changes in microbial community structure and rates of enzyme production with temperature, as indicated by Dalias et al. (2001) and Waldrop and Firestone (2004), or by a greater Al-toxicity effect to microbial communities at low temperatures. It must be noted that SOM decomposition rates depend on microbial “functional diversity” rather than taxonomic diversity (Nannipieri et al. 2003; Bonkowski and Roy 2005; Chaparro et al. 2012). Based on ideas of these previous studies, increase in microbial activity, enzyme production rates, and microbial functional diversity may be higher at higher than at lower temperature.
Heterogeneous distribution of both microbes and organic S fractions in aggregates can also be noteworthy. The large particle size fractions contained higher levels of fungal biomass than the smaller particles (Chiu et al. 2006), whereas 90 % of heterotrophic bacteria preferentially located in the inner part of aggregates (Ranjard et al. 2000). If this is the case, ester sulfate-S may be located in the inner part of the structures where bacteria are predominantly located, whereas C-bonded S may be distributed in the outer part of the macroaggregates where higher abundances of fungi are expected than in the inner part. Fungal activity is often lower than bacterial activity at high temperature (e.g., Pietikäinen et al. 2005). Thus, it is possible that a larger amount of ester sulfate-S was mineralized by bacteria in the inner part of the hierarchical aggregate structures at 35 °C than 20 °C.
In addition, activities of enzymes interacting with minerals and/or organo–mineral complexes vary widely with the temperature (Rao et al. 2000; Kelleher et al. 2004). If enzymes activities mineralized ester sulfate-S have different optimal temperature than those for degradation of C-bonded S, the relationships between the transformation/mineralization rates of these two types of organic S fractions and temperature might also differ.
Conclusions
We performed an incubation experiment to estimate the sequential transformation rates of C-bonded S to ester sulfate-S (velocity 1, v 1) and of ester sulfate-S to sulfate-S (velocity 2, v 2) and the stabilization of ester sulfate-S in volcanic soils. By separating the sequential transformation rates, we showed that the transformation rate of C-bonded S and mineralization of ester sulfate-S depended on temperature and site-specific soil properties; v 2 was significantly (p < 0.05) correlated with active Al concentrations when soils were incubated at 20 °C, and it was sensitive to incubation temperatures. Thus, mineralization of ester sulfate-S would be governed by soil mineral properties and climate. Velocity 1 was significantly (p < 0.01) positively correlated with soil pH at both temperatures, suggesting that a decrease in soil pH suppress the degradation of C-bonded S. Further studies are needed for evaluating the distribution of both ester sulfate-S and C-bonded S in aggregates by using nondestructive methods, and studying ester sulfate-mineral occlusion in soil aggregates and/or adsorption onto mineral surface. In addition, the study of enzyme structures and their association with soil colloids can help to better understand S turnover in volcanic soils.
References
Andriesse JP (1979) A study into the mobility of iron in podzolic Sarawak upland soils by means of selective iron extractions. Neth J Agnc Sci 27:1–12
Badri DV, Weir TL, van der Lelie D, Vivanco JM (2009) Rhizosphere chemical dialogues: plant–microbe interactions. Curr Opin Biotechnol 20:642–650
Bettany JR, Stewart JWB, Saggar S (1979) The nature and forms of sulfur in organic matter fractions of soils selected along an environmental gradient. Soil Sci Soc Am J 43:981–985
Bilen S, Dick W (2011) Sulfite oxidase enzyme activity in soil. Biol Fertil Soils 47:647–654
Bonkowski M, Roy J (2005) Soil microbial diversity and soil functioning affect competition among grasses in experimental microcosms. Oecologia 143:232–240
Boye K, Nilsson I, Eriksen J (2009) Net sulfur mineralization potential in Swedish arable soils in relation to long-term treatment history and soil properties. Biol Fertil Soils 45:743–751
Castellano SD, Dick RP (1990) Cropping and sulfur fertilization influence on sulfur transformations in soil. Soil Sci Soc Am J 55:114–121
Chaparro JM, Sheflin AM, Manter DK, Vivanco JM (2012) Manipulating the soil microbiome to increase soil health and plant fertility. Biol Fertil Soils 48:489–499
Chiu CY, Chen TTH, Imberger K, Tian G (2006) Particle size fractionation of fungal and bacterial biomass in subalpine grassland and forest soils. Geoderma 130:265–271
Churka Blum S, Lehmann J, Solomon D, Caires EF, Alleoni LRF (2013) Sulfur forms in organic substrates affecting S mineralization in soil. Geoderma 200–201:156–164
Dalias P, Anderson JM, Bottner P, CouÃteaux M-M (2001) Long-term effects of temperature on carbon mineralisation processes. Soil Biol Biochem 33:1049–1057
Ellert BH, Bettany JR (1992) Temperature dependence of net nitrogen and sulfur mineralization. Soil Sci Soc Am J 56:1133–1141
Eriksen J (2005) Gross sulphur mineralization-immobilization turnover in soil amended with plant residue. Soil Biol Biochem 37:2216–2224
Eriksen J (2008) Soil sulfur cycling in temperate agricultural systems. In: Jez J (ed) Sulfur: A missing link between soils, crops and nutrition. Agronomy Monograph 50, Madison, WI, pp 25–44
Eriksen J, Murphy MD, Schung E (1998) The soil sulphur cycle. In: Schung E (ed) Sulphur in agroecosystems. Kluwer Academic, Dordrecht, Netherlands, pp 39–73
Fitzgerald JW, Hale DD, Swank WT (1988) Sulphur-containing amino acid metabolism in surface horizons of a hardwood forest. Soil Biol Biochem 20:825–831
Freney JR (1961) Some observations on the nature of organic sulphur compounds in soil. Aust J Agric Res 12:424–432
Freney JR (1967) Sulfur-containing organics. In: McLaren AD, Peterson GH (eds) Soil Biochemistry, vol 1. Marcel Dekker-Inc, New York, pp 220–259
Freney JR (1986) Forms and reactions of organic sulphur compounds in soils. In: Tabatabai MA (ed) Sulphur in agriculture. Soil Science Society of America, Madison, WI, pp 207–232
García-Rodeja E, Nóvoa JC, Pontevedra X, Martínez-Cortizas A, Buurman P (2004) Aluminum fractionation of European volcanic soils by selective dissolution techniques. Catena 56:155–183
Ghani A, McLaren RG, Swift RS (1991) Sulphur mineralization in some New Zealand soils. Biol Fertil Soils 11:68–74
Ghani A, McLaren RG, Swift RS (1992) Sulphur mineralization and transformations in soils as influenced by additions of carbon, nitrogen and sulphur. Soil Biol Biochem 24:331–341
Haynes RJ (1986) Mineral nitrogen in the plant–soil system. Academic, Orlando
Higashi T, Shinagawa A (1981) Comparison of sodium hydroxide-tetraborate and sodium pyrophosphate as extractants of Al/Fe-humus “complexes” in Dystrandepts, Japan. Geoderma 25:285–292
Igwe CA, Zarei M, Stahr K (2009) Colloidal stability in some tropical soils of southeastern Nigeria as affected by iron and aluminum oxides. Catena 77:232–237
International Soil Reference and Information Center (ISRIC) (1993) Procedures for soil analysis, 4th edn. Wageningen, the Netherlands
Johnson CM, Nishita H (1952) Microestimation of sulphur in plant materials, soils, and irrigation waters. Anal Chem 24:736–742
Kaiser K, Zech W (1996) Defects in estimation of aluminum in humus complexes of podzolic soils by pyrophosphate extraction. Soil Sci 161:452–458
Keer JI, McLaren RG, Swift RS (1990) Acetylacetone extraction of soil organic sulphur and fractionation using gel chromatography. Soil Biol Biochem 22:97–104
Kelleher BP, Simpson AJ, Willeford KO, Simpson MJ, Stout R, Rafferty A, Kingery WL (2004) Acid phosphatase interactions with organo–mineral complexes: influence on catalytic activity. Biogeochemistry 71:285–297
Kertesz MA, Mirleau P (2004) The role of soil microbes in plant sulphur nutrition. J Exp Bot 55:1939–1945
Knights JS, Zhao FJ, McGrath SP, Magan N (2001) Long-term effects of land use and fertiliser treatments on sulphur transformations in soils from the Broadbalk experiment. Soil Biol Biochem 33:1797–1804
Kowalenko CG (1993) Extraction of available sulfur. In: Carter MR (ed) Soil sampling and methods of analysis. Lewis, Boca Raton, FL, pp 65–74
Lou G, Warman PR (1992) Labile ester sulphate in organic matter extracted from podzolic soils. Biol Fertil Soils 14:267–271
Lützow MV, Kögel-Knabner I (2009) Temperature sensitivity of soil organic matter decomposition—what do we know? Biol Fertil Soils 46:1–15
Matsuda Y, Sugiyama F, Nakanishi K, Ito S (2006) Effects of sodium chloride on growth of ectomycorrhizal fungal isolates in culture. Mycoscience 47:212–217
Matus F, Garrido E, Sepúlveda N, Cárcamo I, Panichini M, Zagal E (2008) Relationship between extractable Al and organic C in volcanic soils of Chile. Geoderma 148:180–188
Maynard DG, Kalra YP, Radford FG (1987) Extraction and determination of sulfur in organic horizons of forest soils. Soil Sci Soc Am J 51:801–806
McGill WB, Cole CV (1981) Comparative aspects of cycling of organic C, N, S and P through soil organic matter. Geoderma 26:267–286
McKeague JA (1967) An evaluation of 0.1 M pyrophosphate and pyrophosphate dithionite in comparison with oxalate as extractants of the accumulation products in Podzols and some other soils. Can J Soil Sci 47:95–99
McLaren RG, Keer JJ, Swift RW (1985) Sulphur transformations in soils using sulphur-35 labelling. Soil Biol Biochem 17:73–79
Mitchell MJ, David MB, Harrison RB (1992) Sulphur dynamics of forest ecosystems. In: Howarth RW, Stewart JWB, Ivanov MV (eds) Sulphur cycling on the continents, Scope 48. John Wiley & Sons, Chichester, UK, pp 215–254
Mummey D, Holben W, Six J, Stahl P (2006) Spatial stratification of soil bacterial populations in aggregates of diverse soils. Microbial Ecol 51:404–411
Nannipieri P, Ceccanti B, Bianchi D (1988) Characterization of humus–phosphatase complexes extracted from soil. Soil Biol Biochem 20:683–691
Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G (2003) Microbial diversity and soil functions. Eur J Soil Sci 54:655–670
Nelson DW, Sommers LE (1996) Total carbon, organic carbon, and organic matter. In: Sparks DL (ed) Methods of soil analysis, Part 3, Chemical Methods. Soil Sci Soc Am Book Series 5, Madison, WI, pp 961–1010
Nguyen ML, Goh KM (1992) Sulphur mineralization and release of soluble organic sulphur from camp and non-camp soils of grazed pastures receiving long-term superphosphate applications. Biol Fertil Soils 14:272–279
Norman AL, Giesemann A, Krouse HR, Jager HJ (2002) Sulphur isotope fractionation during sulphur mineralization: results of an incubation-extraction experiment with a black forest soil. Soil Biol Biochem 34:1425–1438
O'Brien SL, Jastrow JD (2013) Physical and chemical protection in hierarchical soil aggregates regulates soil carbon and nitrogen recovery in restored perennial grasslands. Soil Biol Biochem 61:1–13
Pietikäinen J, Pettersson M, Bååth E (2005) Comparison of temperature effects on soil respiration and bacterial and fungal growth rates. FEMS Microbiol Ecol 52:49–58
Pietri JCA, Brookes PC (2008) Relationships between soil pH and microbial properties in a UK arable soil. Soil Biol Biochem 40:1856–1861
Pirela HJ, Tabatabai MA (1988) Sulfur mineralization rates and potential of soils. Biol Fertil Soils 6:26–32
Prietzel J, Thieme J, Salomé M, Knicker H (2007) Sulfur K-edge XANES spectroscopy reveals differences in sulfur speciation of bulk soils, humic acid, fulvic acid, and particle size separates. Soil Biol Biochem 39:877–890
Ranjard L, Poly F, Combrisson J, Richaume A, Gourbie're F, Thioulouse J, Nazaret S (2000) Heterogeneous cell density and genetic structure of bacterial pools associated with various soil microenvironments as determined by enumeration and DNA fingerprinting approach (RISA). Microb Ecol 39:263–272
Rao MA, Violante A, Gianfreda L (2000) Interaction of acid phosphatase with clays, organic molecules and organo–mineral complexes: kinetics and stability. Soil Biol Biochem 32:1007–1014
Rasmussen C, Southard RJ, Horwath W (2006) Mineral control of organic carbon mineralization in a range of temperate conifer forest soils. Global Change Biology 12:834–847
Schroth AW, Bostick BC, Graham M, Kaste JM, Mitchell MJ, Friedland AJ (2007) Sulfur species behavior in soil organic matter during decomposition. J Geophys Res 112, G04011. doi:10.1029/2007JG000538
Schuppli PA, Ross GJ, McKeague JA (1983) The effective removal of suspended materials from pyrophosphate extracts of soils from tropical and temperate regions. Soil Sci Soc Am J 47:1026–1036
Soil Survey Staff (2010) Keys to soil taxonomy, 11th edn. USDA, Natural Resources Conservation Service, Washington, DC
Sollins P, Homann P, Caldwell BA (1996) Stabilization and destabilization of soil organic matter: mechanisms and controls. Geoderma 74:65–105
Sollins P, Swanston C, Kleber M, Filley T, Kramer M, Crow S, Caldwell MA, Lajtha K, Bowden R (2006) Organic C and N stabilization in a forest soil: evidence from sequential density fractionation. Soil Biol Biochem 38:3313–3324
Solomon D, Lehmann J, Tekalign M, Fritzsche F, Zech W (2001) Sulfur fractions in particle-size separates of the sub-humid Ethiopian highlands as influenced by land use changes. Geoderma 102:41–59
Solomon D, Lehmann J, Martinez CE (2003) Sulfur K-edge XANES spectroscopy as a tool for understanding sulfur dynamics in soil organic matter. Soil Sci Soc Am J 67:1721–1731
Solomon D, Lehmann J, Zarruk KK, Dathe J, Kinyangi J, Liang B, Machado S (2011) Speciation and long- and short-term molecular-level dynamics of soil organic sulfur studied by X-ray absorption near-edge structure spectroscopy. J Environ Qual 40:704–718
Stevenson FJ, Cole MA (1999) Cycles of soil—carbon, nitrogen, phosphorus, sulfur, micronutrients, 2nd edn. John Wiley & Sons, Chichester
Tabatabai MA (1996) Sulphur. In: Sparks DL (ed) Methods of soil analysis, Part 3, Chemical Methods, book series 5. Soil Sci Soc Am, Madison, WI, pp 921–960
Tabatabai MA, Al-Khafaji AA (1980) Comparison of nitrogen and sulfur mineralization in soils. Soil Sci Soc Am J 44:1000–1006
Tanikawa T, Takahashi M, Imaya A, Ishizuka K (2009a) Highly accumulated sulfur constituents and their mineralogical relationships in Andisols from central Japan. Geoderma 151:42–49
Tanikawa T, Takahashi M, Noguchi K, Shigenaga H, Nagakura J, Sakai H, Ishizuka K, Akama A (2009b) Nutritional properties of trees and soils in Oku-Nikko Region, central Japan—comparisons with the other subalpine regions. Environ Sci 22:401–414 (in Japanese with English abstract)
Tanikawa T, Yamashita N, Aizawa S, Ohnuki Y, Yoshinaga S, Takahashi M (2013) Soil sulfur content and its spatial distribution in a small catchment covered by volcanic soil in the montane zone of central Japan. Geoderma 197–198:1–8
Tisdall JM, Oades JM (1982) Organic matter and water stable aggregates in soils. J Soil Sci 33:141–163
Wada K (1989) Allophane and imogolite. In: Dixon JB, Weed SB (eds) Minerals in soil environment. SSSA, Madison, WI, pp 1051–1087
Waldrop MP, Firestone MK (2004) Altered utilization patterns of young and old soil C by microorganisms caused by temperature shifts and N additions. Biogeochemistry 67:235–248
Wang J, Solomon D, Lehmann J, Zhang X, Amelung W (2006) Soil organic sulfur forms and dynamics in the Great Plains of North America as influenced by long-term cultivation and climate. Geoderma 133:160–172
Williams CH (1967) Some factors affecting the mineralization of organic sulphur in soils. Plant Soil 26:205–223
Yagasaki Y, Mulder J, Okazaki M (2006) Comparing the activity of aluminum in the B horizons developed from volcanic ash deposits in Japan, dominated by short-range ordered aluminosilicates and crystalline clay minerals, respectively. Geochim Cosmochim Acta 70:147–163
Yoshinaga N (1986) Clay minerals. In: Wada K (ed) Ando soils in Japan. Kyushu university press, Fukuoka, Japan, pp 41–56
Zhao FJ, Lehmann J, Solomon D, Fox MA, McGrath SP (2006) Sulphur speciation and turnover in soils: evidence from sulphur K-edge XANES spectroscopy and isotope dilution studies. Soil Biol Biochem 38:1000–1007
Acknowledgments
We thank Dr. A. Imaya, Dr. Y. Inagaki, Dr. K. Ono, and the other members of the Department of Forest Site Environment of the Forestry and Forest Products Research Institute; Dr. Y. Matsuda (Mie University), Dr. Y. Hirano (Nagoya University) for making a number of helpful suggestions. We also thank Prof. J. Prietzel (Technische Universität München), Dr. R. Wagai, Dr. S. Hiradate, and Dr. M. Asano (National Institute for Agro-Environmental Sciences) for invaluable suggestions concerning soil organic S stabilization. We additionally thank Dr. P. Nannipieri, the editor-in-chief, and two anonymous reviewers for their critical comments on an earlier draft of the manuscript. We are grateful for the financial support of the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT, 21780162, 24580096); the Ministry of Agriculture, Forestry and Fisheries, Japan; and a Grant for Environmental Research Projects from the Sumitomo Foundation (no. 103331). This work was also supported by the Program for Supporting Activities for Female Researchers funded by MEXT's Special Coordination Fund for Promoting Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kenichi Nakanishi Deceased
Rights and permissions
About this article
Cite this article
Tanikawa, T., Noguchi, K., Nakanishi, K. et al. Sequential transformation rates of soil organic sulfur fractions in two-step mineralization process. Biol Fertil Soils 50, 225–237 (2014). https://doi.org/10.1007/s00374-013-0849-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-013-0849-8