
Abstract
Purpose
We aimed to determine if family history (FH) of prostate cancer (PC) influenced cancer control after radical prostatectomy (RP).
Methods
Patients were evaluated in a prospectively-collected PC family database: The focus was on hereditary prostate cancer (HPC) defined by Johns Hopkins criteria and sporadic prostate cancer (SPC), rigorously defined by absence of prostate cancer in ≥ 2 brothers aged ≥ 60 years. Additionally, patients with first-degree (FPC) and non-first-degree PC (non-FPC) were assessed. Endpoints were biochemical recurrence-free survival (BRFS) and prostate cancer-specific survival (CSS). Finally, clinico-pathological characteristics were compared and multiple proportional hazards regression was used to identify prognostic factors.
Results
In total 11,654 patients were included (807 HPC, 2251 FPC, 8072 non-FPC and 524 SPC). Familial imposition (HPC/FPC) was associated with a younger age at diagnosis. Thus, HPC patients were diagnosed 2.9 years earlier than SPC patients with more locally advanced tumors (≥ pT3). With a median follow up of 6.2 years (range 0–31.5) BRFS was significantly different when stratified by FH. In pairwise analyses BRFS differed significantly for HPC compared to SPC (HR = 1.27). Consecutively FH was identified as prognostic factor for BRFS (p = 0.021) together with age, PSA, pathologic characteristics and adjuvant androgen deprivation. Analyses of CSS did not show a difference.
Conclusion
Patients with FH of PC are likely to be diagnosed earlier and present a higher proportion of locally advanced disease. In addition, men with FH are at higher risk of biochemical recurrence after surgery but reveal similar outcomes regarding prostate cancer-specific survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Family history (FH) of prostate cancer (PC) is present in up to 20% of PC patients [1, 2]. Hereditary PC (HPC) is a subgroup of familial PC with a likelihood of genetic predisposition [3,4,5,6]. Using the Johns Hopkins criteria for HPC, approximately 2.6% of all PC patients and 11.5% of patients with FH are considered to have HPC [7]. While it is generally agreed that FH is associated with an increased risk of disease and an earlier age of diagnosis, it is less clear whether HPC confers worse outcomes when compared to sporadic PC (SPC) [1, 2, 8].
The impact of FH on prostate cancer-specific survival (CSS) has been studied revealing conflicting results, likely due to lack of sufficient follow-up [1, 8,9,10,11,12,13]. While most studies have found no association between FH and biochemical recurrence-free survival (BRFS) after curative treatment [8, 11,12,13]. Lee et al. found improved disease-free survival in patients without FH [14]. Contrary to this, Kupelian et al. described lower BRFS for patients with familial PC treated in the year ≤ 1992 compared to SPC patients. Regarding patients treated in the year ≥ 1993 no difference in BRFS occurred [15].
A challenge to this body of literature is the use of different definitions of FH and inclusion criteria. Some studies have included patients with fewer than 2 affected relatives while others used the strict Johns Hopkins criteria [3]. In addition, the current definition of SPC may import misinterpretations, since patients are deemed as SPC despite absence of a complete medical history of first-degree relatives and thus potentially including unrealized cases of HPC. In order to reduce these sources of error we selected patients using strict criteria for both SPC- and HPC patients. SPC patients were defined with ≥ 2 brothers aged at least 60 years and negative anamneses for PC. This definition is even stricter than the definition Valeri et al. used in their study (2 + non-affected brothers at least 50 years old) [16].
In the current study, a prospective German family registry was employed; this registry prospectively employed the strict definitions of HPC and SPC for stratifying men with PC who underwent RP. As a result, it provides an unbiased evaluation of the difference in clinical outcomes for men with HPC versus SPC. Despite focusing mainly on the comparison of HPC vs. SPC, we additionally evaluated patients with familial first-degree (FPC) and non-first-degree prostate cancer (non-FPC) in order to provide a complete outline of the familial spectrum.
Patients and methods
Participants
Patients who underwent RP as a curative treatment for PC between 1994 and 2010 were selected from the German database Familial Prostate Cancer; the project was established in 1992 and currently includes 38,552 cases within 29,732 families. The prospective, multi-center database has been previously described [8, 17]. Briefly, participating clinics and urologists recruited all patients diagnosed with PC and invited them to complete demographic, clinical and family history questionnaires annually. Self-reported FH of PC was verified by histopathological reports of patients and affected relatives. Relatives subsequently diagnosed with PC were included as they were identified during annual follow-up. Patients were classified according to the strict inclusion criteria for this study: SPC patients were defined as ≥ 2 brothers aged at least 60 years and no FH of PC, HPC patients were defined according to the Johns Hopkins criteria [3], with at least one of the following criteria: ≥ 3 affected relatives within any nuclear family (father and 2 brothers; group of 3 brothers), occurrence of PC in each of 3 generations in either the proband’s paternal or maternal lineage, or 2 relatives affected at < 55 years, FPC patients were defined as clustering of at least two first-degree relatives with PC and non-FPC was defined as having no first-degree relative with PC in the family. For all patients and affected family members, histopathological confirmed PC was required. Figure 1 shows the steps for identifying the 4 groups of patients according to their family history.

Study design. SPC sporadic prostate cancer, HPC hereditary prostate cancer, FPC first-degree prostate cancer and non-FPC non-first-degree prostate cancer
All patients had clinically localized PC treated by RP, including lymph node dissection as appropriate. Adjuvant radiation therapy and androgen deprivation were employed, generally based on pathologic stage. Pathologic specimens were centrally reclassified according to the “Union international contre le cancer” version 2002 for standardizing reports, using the pathology report of the individual clinic. Pathological staging included local tumor extension, lymph node involvement, Gleason Score and grading according to WHO-guidelines (GI-III). As Gleason grading was not assessed routinely until the late nineties, tumors for patients initially included in the database were classified as well, moderately and poorly differentiated (GI, GII and GIII, respectively). Following RP, patients received annual questionnaires that included updated information on PC, FH, PSA, and additional therapies, such as androgen deprivation or radiotherapy. Biochemical failure was defined as two successive PSA measurements ≥ 0.2 ng/ml [18].
Statistics
For quantitative variables, mean, median, minimum and maximum values were calculated while percentages were given for qualitative data. Chi-squared tests were used to compare subgroups.
BRFS was defined as time from RP to biochemical recurrence and CSS by a PC related death. For BRFS and CSS, Kaplan–Meier curves were computed along with 95% CI for 10-year survival rates. To assess the prognostic value of individual clinical characteristics for both BRFS and CSS in the total population of subjects with SPC, FPC, non FPC and HPC, first separate proportional hazards models were fitted, and then multiple proportional hazards regression with backward elimination was performed (selection level 5%). Hazard ratio with 95% confidence interval and p value were calculated. Statistical analyses were performed with SAS version 8.2 (SAS Institute Inc., Cary, NC, USA). All statistical tests were performed at the two-sided 0.05 significance level.
Results
From the German Familial Cancer Database 11,654 patients met the study inclusion criteria, based on a complete family history and follow up; 807 patients with HPC, 2251 patients with FPC, 8072 patients with non-FPC and 524 patients with SPC (Fig. 1). The HPC cohort included 40 patients meeting the criteria of 2 or more affected relatives with an onset age of 55 years or less, 1 patient with 3 or more affected relatives from 3 generations, 1 patient with 3 or more affected relatives from 3 generations and additionally 3 or more first-degree relatives in the nuclear family, while the majority (n = 765) had 3 or more affected first-degree relatives.
All selected patients were Caucasian and had questionnaire-based follow-up of a median of 6.2 years (range 0–31.5 years). Of these patients, 91.2% (n = 10,308) underwent concurrent pelvic lymph node dissection at the time of RP.
Clinical characteristics and treatment at diagnosis
Clinical characteristics of the enrolled patients are summarized in Table 1. The median age at diagnosis was 64.5 years in the complete cohort. In overall analyses, there was a significant difference between age at diagnosis across FH subgroups. Focusing on HPC and SPC patients as the extreme ends of the family history spectrum, median age at diagnosis was 63.2 years in HPC (range 35.7–77.7 years) vs. 66.1 years in SPC (50.0–82.1 years). Thus, comparing medians, HPC patients were diagnosed 2.9 years earlier than SPC patients. Notably, early onset PC (≤ 55 years) was observed more frequently in familial-associated PC patients: 13.4% of HPC, 11.7% of FPC, 8.7% of non-FPC, and finally only 2.5% of those with SPC.
Evaluation of the pathologic stage revealed a somewhat higher rate of locally advanced tumors (≥ pT3) in HPC patients, 38.2%, compared to 33.2% in FPC, 34.6% in non-FPC, and 32.8% in SPC patients. Similarly, further testing for the proportion of patients with organ-confined tumors, defined as ≤ pT2c and pN0 vs. > pT2c or pN1, revealed no significant difference. However, there was a trend for more non organ confined tumors in HPC patients, 39.3%, compared to 34.2% in FPC, 35.7% in non-FPC and 34.4% in SPC patients. There was a significant difference in the rate of lymph node metastases between FH subgroups. With respect to tumor grading, overall analyses displayed a significant difference, with the lowest proportion of well-differentiated tumors (G I) in SPC patients.
Further analyses of the clinical characteristics revealed no difference, with respect to PSA-value at diagnosis, grading and resection margins, or the use of adjuvant treatment modalities including androgen deprivation and radiation therapy.
Clinical outcomes
Median time of follow-up was 9.0 years (range 0.3–23 years) in HPC, 6.6 years (range 0.3–31.5 years) in FPC, 5.8 years (range 0.3–24.3 years) in non-FPC and 6.9 years (0.3–17.8 years) in SPC patients.
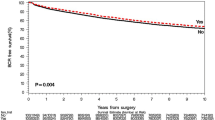
Analyses of BRFS revealed a significant difference when stratified by family history (Fig. 2, Table 2). Pairwise comparison of HPC and SPC patients as the extreme ends of the family history spectrum revealed a HR of 1.27. The 10 year BRFS rate for HPC patients was 53.0% (95% CI 48–57%) compared to 58.7% (95% CI 53–64%) for SPC patients. There was no obvious difference between FPC and non-FPC patients with 60.5% (95% CI 58–63%) and 60.9% (95% CI 60–62%).

Kaplan–Meier curve for biochemical recurrence free survival (BRFS) stratified by family history. HPC hereditary prostate cancer, FPC first-degree prostate cancer, non-FPC non first-degree prostate cancer, SPC sporadic prostate cancer, CI confidence interval
The prognostic value of each potential prognostic factor for BRFS was assessed in separate proportional hazards regression models, and for selection of important prognostic factors, multiple regression with backward elimination was performed (Table 2). Results confirmed FH as a prognostic factor for BRFS (p = 0.021) along with age, PSA, pathologic stage and node stage, surgical margins, tumor grade and adjuvant AD (Table 2).
In contrast, analyses of CSS showed no difference across FH subgroups (p = 0.148) (Fig. 2, Table 3). Pairwise comparison of HPC and SPC patients revealed no difference in CSS. The missing prognostic value of FH for CSS was confirmed by multiple proportional hazards regression where only clinical and histopathological characteristics were identified as prognostic factors (Table 3) (see Fig. 3).

Kaplan–Meier curve for prostate cancer-specific survival (CSS) stratified by family history. HPC hereditary prostate cancer; FPC first-degree prostate cancer, non-FPC non first-degree prostate cancer, SPC sporadic prostate cancer, CI confidence interval
Discussion
By applying a rigorous definition of SPC and HPC to a medically verified comprehensive familial registry, this study represents the first of its kind to assess in an unbiased manner whether clinical outcomes following RP are affected by FH of PC. The advantages of this analysis compared to previous studies are the strict definitions of FH leading to a most discriminative population as well as rigorous pathologic confirmation of disease status for both patient and family members. For patients with less-extensive FH, it would be expected that outcomes would be intermediate, somewhere between HPC and SPC. In order to provide a complete outline of the familial spectrum we additionally evaluated patients with first-degree (FPC) and non-first-degree PC (non-FPC) although our main focus was to examine the extreme ends of the family history spectrum with HPC and SPC patients. In this analysis, we found that FH did not affect CSS after radical prostatectomy. This observation, combined with a marginal reduction of the 10 year BRFS rate for HPC patients of 53.0% compared to 58.7% for SPC patients, suggests that FH has a minor impact on disease outcomes after RP, although FH was selected as an important prognostic factor in multiple proportional hazards regression.
The medical literature is replete with studies examining the impact of FH on the risk of PC. In the Finnish Prostate Cancer Screening Trial of 80,144 subjects, 31,866 were randomized to the screening arm. A total of 1723 (7.3%) reported at least one first-degree relative with prostate cancer. While the risk of low-grade cancer was increased in men with a FH of PC (RR 1.46), the risk of high-grade cancer was reduced (RR 0.48). A challenge to the seemingly paradoxical conclusion of this study regarding high grade disease was the very small number of Gleason 8–10 tumors (n = 9) among patients with a FH of PC [10]. In the PLCO trial conducted by the National Cancer Institute (U.S.) involving 65,179 men, 7314 (11.2%) were diagnosed with prostate cancer; FH was assessed with a baseline questionnaire. A FH of PC was associated with a higher incidence of PC (16.9 vs 10.8%) and higher prostate cancer-specific mortality (PCSM) (0.56 vs 0.37%, both comparisons p < 0.01). Interestingly, while the impact of screening on mortality in the overall study was negative, among men with a FH of prostate cancer, screening was associated with decrease in PCSM [19].
In a study of 1711 men undergoing radiotherapy, FH of PC was not associated with a greater risk of disease recurrence. However, if only men with two or more first-degree relatives were considered, they had a higher risk of biochemical failure and distant metastases although PCSM and overall mortality was not different. Like other single-institution studies in which the authors do not report whether a standardized assessment of FH was prospectively employed, the accuracy of FH data in this and other studies cannot be assessed [20]. In a study of 3560 subjects undergoing RP at the Mayo Clinic, 865 were categorized as having familial PC while 133 with HPC. PSA was generally higher in those with HPC but 10-year cancer-specific outcomes were no different among the groups. In this study, it is notable that FH assessment was conducted through a survey mailed at one time (March 1995) to 4616 patients in their PC database; only those men who responded were included in this study. Concerns that arise from such a technique include censoring of patients who died between 1987 and 1995 including possibly those who died of PC, accuracy of self-reporting, as well as differential responses to the questionnaire based on FH of PC [12].
In a study of 557 men undergoing RP from 1989 to 2000, biochemical recurrence was higher in the non-FH group while other outcomes were similar. The authors postulated that lower biochemical recurrence in the FH group was due to heightened vigilance for screening and an earlier diagnosis. In this retrospective study, chart reviews were used to assess FH and the authors acknowledge that data were not consistently available [14]. Among 720 patients with PC treated with RP between 1987 and 1996, with a median follow-up of only 30 months, 12% had positive FH. 5-year BRFS was worse with FH (46%) compared with those without FH (66%). The authors acknowledged, like other series, that data were obtained from chart reviews and that they were concurrently collecting confirmatory data (e.g., pathology reports) to validate FH [21]. Finally, a study of 481,011 men at enrollment without cancer in 1982 followed for 9 years found 1922 deaths from PC. FH data were collected through a baseline questionnaire. FH was related to risk of fatal PC (HR 1.6). If two or more affected relatives were affected RR was 3.19. If relatives were diagnosed before age 65, RR was 2.03 [22].
The current study, with an intermediate duration of follow-up, conducted during a more recent period of PC testing and treatment, has a number of strengths when compared with previous examinations of the relationship of FH and PC risk. A primary purpose of this study was related to FH and thus, prospectively, FH was assessed. Additionally, FH of PC was validated using pathology reports. As family members, especially at older ages, often do not relate accurately their diagnoses to offspring or siblings, the precise diagnosis can be misattributed. All patients were also followed prospectively with close attention to collection of PC outcomes, minimizing risk of those lost to follow-up.
We acknowledge that there are likely to be inherent differences in screening and treatment of patients with PC based on their FH of PC. For example, a man with FH may be more likely to undergo PSA testing at a younger age, potentially leading to a greater risk of diagnosis of low-grade cancer as was seen in the current study [23]. Such a man may be more likely, as well, to have a recommendation for biopsy at a lower PSA value and to undergo more frequent biopsies; he is probably also more likely to accept a biopsy recommendation at any level of PSA [24]. These biases likely increase the risk of prostate cancer detection, in addition to skewing the detection to lower-grade tumors.
As it has been demonstrated that, for men with higher-risk categories of PC, treatment may reduce risk of metastases and death, other biases may affect longer-term and more important outcomes of PC such as metastases and PC death [25, 26]. If men with a FH of PC face a PC diagnosis, there is the possibility that they and their physicians may treat these tumors more intensively, potentially leading to a perceived lower risk of these important disease-related outcomes. It is important to recognize that all of these biases were likely operational in the current study and, short of a randomized trial that mandates precise screening, biopsy, and treatment, no evaluation of this relationship can fully avoid this major sources of bias.
Our data strongly suggest that, in a contemporary screening and treatment environment, FH of PC may slightly increase the risk of PSA recurrence after RP but with current management, FH does not have a significant impact on risk of death from PC. We cannot rule out the possibility that patients with a FH of PC should have a different treatment planning approach to their tumors based on these data but only that current medical treatments appear to erase any differences in disease prognosis in these men. Knowledge about the mutation of high risk genes or single nucleotide polymorphisms (SNPs) related to an increased risk of PC and/or a more aggressive course of the disease would have been interesting but was not available in our patient population.
Conclusion
This large-scale study, using a rigorous definition of family history of prostate cancer, suggests that men with hereditary prostate cancer are diagnosed at a younger age, present with a higher disease stage, and have a higher risk of PSA recurrence after surgical therapy. Nonetheless, with the employment of current therapies, these risks do not translate to a higher risk of prostate cancer death.
References
Hemminki K (2012) Familial risk and familial survival in prostate cancer. World J Urol 30(2):143–148
Heidenreich A, Bastian PJ, Bellmunt J et al (2014) EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and local treatment with curative intent—update 2013. Eur Urol 65(1):124–137
Carter B, Bova G, Beaty T et al (1993) Hereditary prostate cancer: epidemiologic and clinical features. J Urol 150(3):797–802
Grill S, Fallah M, Leach RJ et al (2015) Incorporation of detailed family history from the swedish family cancer database into the PCPT risk calculator. J Urol 193(2):460–465
Karyadi DM, Zhao S, He Q et al (2015) Confirmation of genetic variants associated with lethal prostate cancer in a cohort of men from hereditary prostate cancer families. Int J Cancer 136(9):2166–2171
Maier C, Herkommer K, Luedeke M, Rinckleb A, Schrader M, Vogel W (2014) Subgroups of familial and aggressive prostate cancer with considerable frequencies of BRCA2 mutations. Prostate 74(14):1444–1451
Herkommer K, Schmidt C, Gschwend J (2011) Ten years national research project “familial prostate cancer”: problems in identifying risk families. Urol A 50(7):813–820
Heck MM, Kron M, Gschwend JE, Herkommer K (2012) Effect of family history on outcome in German patients treated with radical prostatectomy for clinically localised prostate cancer. Eur J Cancer 48(9):1312–1317
Pakkanen S, Kujala PM, Ha N, Matikainen MP, Schleutker J, Tammela TL (2012) Clinical and histopathological characteristics of familial prostate cancer in Finland. BJU Int 109(4):557–563
Saarimäki L, Tammela TL, Määttänen L et al (2015) Family history in the finnish prostate cancer screening trial. Int J Cancer 136(9):2172–2177
Sacco E, Prayer-Galetti T, Pinto F et al (2005) Familial and hereditary prostate cancer by definition in an Italian surgical series: clinical features and outcome. Eur Urol 47(6):761–768
Siddiqui SA, Sengupta S, Slezak JM, Bergstralh EJ, Zincke H, Blute ML (2006) Impact of familial and hereditary prostate cancer on cancer specific survival after radical retropubic prostatectomy. J Urol 176(3):1118–1121
Rouprêt M, Fromont G, Bitker M-O, Gattegno B, Vallancien G, Cussenot O (2006) Outcome after radical prostatectomy in young men with or without a family history of prostate cancer. Urology 67(5):1028–1032
Lee KL, Marotte JB, Ferrari MK, McNeal JE, Brooks JD, Presti JC Jr (2005) Positive family history of prostate cancer not associated with worse outcomes after radical prostatectomy. Urology 65(2):311–315
Kupelian PA, Reddy CA, Reuther AM, Mahadevan A, Ciezki JP, Klein EA (2006) Aggressiveness of familial prostate cancer. J Clin Oncol 24(21):3445–3450
Valeri A, Azzouzi R, Drelon E et al (2000) Early-onset hereditary prostate cancer is not associated with specific clinical and biological features. Prostate 45(1):66–71
Paiss T, Wörner S, Kurtz F et al (2003) Linkage of aggressive prostate cancer to chromosome 7q31-33 in German prostate cancer families. Eur J Hum Genet 11(1):17–22
Heidenreich A, Bastian PJ, Bellmunt J et al (2014) EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol 65(2):467–479
Liss MA, Chen H, Hemal S et al (2015) Impact of family history on prostate cancer mortality in white men undergoing prostate specific antigen based screening. J Urol 193(1):75–79
Bagshaw H, Ruth K, Horwitz EM, Chen DY, Buyyounouski MK (2014) Does family history of prostate cancer affect outcomes following radiotherapy? Radiother Oncol 110(2):229–234
Kupelian PA, Klein EA, Witte JS, Kupelian VA, Suh JH (1997) Familial prostate cancer: a different disease? J Urol 158(6):2197–2201
Rodríguez C, Calle EE, Miracle-McMahill HL et al (1997) Family history and risk of fatal prostate cancer. Epidemiology 8(6):653–657
Reed A, Ankerst DP, Pollock BH, Thompson IM, Parekh DJ (2007) Current age and race adjusted prostate specific antigen threshold values delay diagnosis of high grade prostate cancer. J Urol 178(5):1929–1932
Norrish A, McRae C, Cohen R, Jackson R (1999) A population-based study of clinical and pathological prognostic characteristics of men with familial and sporadic prostate cancer. BJU Int 84(3):311–315
D’Amico AV, Chen M-H, Renshaw AA, Loffredo M, Kantoff PW (2008) Androgen suppression and radiation vs radiation alone for prostate cancer: a randomized trial. JAMA 299(3):289–295
Thompson IM, Tangen CM, Paradelo J et al (2009) Adjuvant radiotherapy for pathological T3N0M0 prostate cancer significantly reduces risk of metastases and improves survival: long-term followup of a randomized clinical trial. J Urol 181(3):956–996
Author information
Authors and Affiliations
Contributions
M Thalgott: Manuscript writing, Project development. M Kron: Data analysis, Data management, Manuscript editing. JM Brath: Data collection. DP Ankerst: Data analysis, Manuscript editingIM Thompson: Manuscript editing. JE Gschwend: Protocol development. K Herkommer: Protocol and project development, Manuscript editing.
Corresponding author
Ethics declarations
Funding
No funding was received for this study.
Conflict of interest
The authors declare no potential conflict of interest related to this article.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Thalgott, M., Kron, M., Brath, J.M. et al. Men with family history of prostate cancer have a higher risk of disease recurrence after radical prostatectomy. World J Urol 36, 177–185 (2018). https://doi.org/10.1007/s00345-017-2122-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00345-017-2122-5