
Abstract
Aechmea fasciata is a well-known ornamental flowering plant in the bromeliad family. It is proposed that a small burst of ethylene synthesis in the meristem triggers flowering in pineapple and other bromeliads in response to diverse environmental and endogenous signals. A. fasciata showed an age-dependent response: adult plants were induced to flower successfully under ethylene treatment but juvenile plants did not. To better understand the mechanism of different responses to ethylene in transcriptome and flowering induction by ethylene, we performed a comparative analysis of A. fasciata transcriptome. Four libraries of A. fasciata adult and juvenile plants under water and ethylene treatment were sequenced by Illumina deep sequencing, 55,238,936, 53,797,292, 53,471,812, and 53,485,862 qualified Illumina clean reads, respectively, with 90 bp mean length, respectively. Unigenes of the four libraries were assembled and then merged into an unified library with 86,609 sequences and a mean size of 987 bp. After searching against Nr, KEGG, Swiss-Prot, and COG databases, 28,350 sequences were assigned to 129 KEGG pathways, 28,289 unigenes were categorized into 64 functional groups, and 19,293 sequences were classified into 25 COG categories. Through differential expression analysis, 56 DEGs related to flowering were identified. The critical genes correlated with flowering were selected and confirmed by qRT-PCR analysis. This study provided a global survey of changes in transcriptomes of A. fasciata in response to ethylene. The analyses of transcriptome profiles imply that FT is upregulated in the adult plant and results in flowering. Moreover, the differential expression of GI, DELLA, GAD1, AP2, and so on indicated that a complicated network participated in the induction of flowering by ethylene, which will help in the future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Aechmea fasciata is a well-known tropical flowering plant in the bromeliad family. It is proposed that a small burst of ethylene synthesis in the meristem triggers flowering in pineapple and other bromeliads in response to diverse environmental and endogenous signals (Trusov and Botella 2006). To control flowering time and improve flowering uniformity, ethylene and its alternatives have been used to induce flowering in large-scale industrial bromeliad production for a long time. Under ethylene treatment, A. fasciata showed an age-dependent response: adult plants were induced to flower successfully but juvenile plants were not (Jung and others 2012).
Plants monitor environmental and developmental changes through a sophisticated regulatory network to control the timing of flowering. A network of six major pathways that control flowering time was characterized in Arabidopsis thaliana, including the photoperiod and vernalization pathways in response to seasonal changes of day length and temperature, the ambient temperature pathway in response to daily temperature, and age, autonomous and gibberellin integrated physiological cues of plants (Fornara and others 2010). Recent studies indicated that FlOWERING LOCUS T (FT) expression by various factors including temperature, photoperiod, GA, and plant age was induced by transcriptional activator CONSTANS (CO) and triggered plant flowering (Andres and Coupland 2012). Light signaling and the circadian clock controlled the activity of CO protein (Andres and Coupland 2012). The circadian clock-regulated protein GIGANTEA (GI), a large nuclear protein, plays a key role in regulating CO expression (Song and others 2013). It has been reported that GI can trigger activation of FT directly in a CO-independent manner (Sawa and Kay 2011). The stability of CO protein is also controlled by blue light photoreceptor cryptochromes CRY2, through a tripartite complex formed by CRY2, RING-finger E3 ubiquitin ligase CONSTITUTIVE PHOTOMORPHOGENESIS 1 (COP1) and SUPRESSOR OF PHYA-105 1 (SPA1), repressing the activity of COP1-SPA1 and leading to CO protein stabilization (Zuo and others 2011). GA affected flowering time through the control of DELLA protein stabilization. Bioactive GAs promote binding of the GID1 receptor to DELLA and initiate DELLA degradation (Mutasa-Gottgens and Hedden 2009). DELLA reduces the expression of FT, acting as a negative regulator of the GA signaling pathway and flowering (Galvao and others 2012). DELLA attenuates the binding ability of phytochrome-interacting factor 4 (PIF4), which activates the expression of FT at high temperatures (Kumar and others 2012). DELLA also reduces the level of MicroRNA172 (miR172), delaying flowering partly (Yu and others 2012). APETALA 2 (AP2), TARGET OF EAT 1 (TOE1), TARGET OF EAT 2 (TOE2), and TARGET OF EAT 3 (TOE3), acting as floral repressors, are the targets of miR172 and downregulated by miR172 (Aukerman and Sakai 2003). The abundance of miR172 increases as plants develop, in the mean while, miR156 correlated negatively with the expression level of miR172 (Jung and others 2007). The miR156 targets are SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) transcripts, of which SPL9 activates the transcription of MIR172 and SPL3 binds to promoter of FT to induce FT expression (Wu and others 2009). A few of transcription repressors such as FLOWERING LOCUS C (FLC), SHORT VEGETATIVE PHASE (SVP), and TEMPRANILLO 1 (TEM1) bind to the FT locus (Song and others 2013). FLC plays a key role in vernalization and autonomous pathways, as well as in ABA and BRs mediating flower timing (Fornara and others 2010; Wang and others 2013). It is supposed that the floral repressors including SVP, TEM1, AP2, TOE1, TOE2, and TOE3 regulated by development stages, prevent precocious flowering by repressing FT expression either under unsuitable conditions for flowering or in the juvenile developmental stages (Song and others 2013). After expression in the leaf, FT is transferred to the shoot apical meristem and interacts with bZIP transcription factor FD, promoting the expression of MADS-box factor APEATELA1 (AP1) and initiating flower development.
Ethylene was considered an ‘aging’ hormone often because it promotes such developmental processes as senescence, abscission, and ripening, and it also promotes flowering in Bromeliads but inhibits it in others (Schaller 2012). The essential elements of the ethylene signaling pathway include the ethylene receptors such as ETR1, ERS1, ETR2, ERS2, and EIN4, the transmembrane protein EIN2, the Raf-like kinase CTR1, and the transcription factors EIN3 and EILs. A signal triggered by ethylene is transduced via CTR1 and EIN2 to EIN3/EILs, which are the transcription factors localized in the nucleus and regulate a wide array of plant pathways (Merchante and others 2013). Of the key elements of the ethylene signaling pathway, the ethylene receptors and CTR1 are negative regulators, while EIN3 and EILs act as positive regulators (Hua and Meyerowitz 1998; Kieber and others 1993; Solano and others 1998). Multiple mechanisms for the ethylene-mediated plant developmental response have been identified, in which EILs and the downstream ethylene response DNA-binding factor (EDF) and ethylene response factor (ERF) play a key role (Chuck and Hake 2005; Lingam and others 2011; Schaller 2012; Zhong and others 2009). In A. thaliana, ethylene delays flowering by reducing bioactive GA levels and enhancing DELLA accumulation via EIN3 (Achard and others 2007). But the mechanism for ethylene-induced flowering in bromeliads remains unclear, though ethylene has been used to induce flowering in bromeliads for a long time.
In our previous study, we performed a comparative transcriptome analysis of A. fasciata juvenile and adult plants under ethylene treatment, and many differentially expressed genes (DEGs) related to flower development and ethylene signaling were indentified. However, it is hard to determine whether the differential expression level of DEGs detected above was in response to ethylene or due to the difference of development stages. To further detect the difference between adult plant and juvenile plant responses to ethylene and genes with regard to age-dependent flowering induction of A. fasciata, four transcriptome libraries of adult plants and juvenile plants under ethylene treatment and water treatment were sequenced and analyzed, which showed a meaningful result. Data of this experiment will guide future studies on mechanisms of flowering induction by ethylene in bromeliads.
Methods
Plant Sample Preparation and RNA Isolation
A. fasciata juvenile (6–8 months) and adult (11–14 months) plants were collected from a greenhouse located in the experimental area at the Institute of Tropical Crop Genetic Resources, Chinese Academy of Tropical Agricultural Sciences (CATAS). A. fasciata plants were treated with 400 ul 1−1 of ethrel for 24 h (ambient temperature was 30–32 °C), and plants were also treated with water as a control. The shoot tip tissues and surrounding core leaves were physically isolated and immediately frozen in liquid nitrogen. Frozen tissues from each samples were ground to a fine powder using a mortar and pestle. Total RNA was extracted from the sample for 8 min at 65 °C using the CTAB extraction method. The RNA samples were treated with 10 units of DNaseI (Takara) for 30 min at 37 °C to remove the genomic DNA. The RNA quantity was examined using spectrophotometry, and the RNA quality was checked by gel electrophoresis.
Library Preparation for Transcriptome Analysis and Illumina Deep Sequencing
Using the OligoTex mRNA mini kit (Qiagen), poly-A-containing mRNAs were enriched from the total RNA samples. Then the mRNA was fragmented into small pieces using an RNA fragmentation kit (Ambion). These short fragments were used as the templates for cDNA synthesized. The double-stranded cDNA strand was synthesized using a double-stranded cDNA synthesis kit (Invitrogen). The cDNA fragments were purified using the QiaQuick PCR extraction kit (Qiagen) and eluted with EB buffer. The short cDNA fragments were then liganded with sequencing adapters, and the products were subsequently purified and amplified using PCR to prepare the final cDNA libraries. The four constructed cDNA libraries were then sequenced using Illumina HiSeq™ 2000. The output raw reads were filtered by the trimming of adaptor sequences, empty reads, and ambiguous nucleotides (‘N’ in the end of the reads).
Functional Annotation and Classification
The Clean reads obtained were then assembled using the Trinity de novo transcriptome assembly software (Grabherr and others 2011). The core eukaryotic gene-mapping approach (CEGMA) was used to assess the completeness of the transcriptome assembly. Then, BLASTX alignments (e value < 1e-5) between the unigenes were searched against protein databases, including Nr, Swiss-Prot, KEGG, and COG, and the best matched results were used to determine the sequence direction of the unigenes. Then, the ESTScan software was used to predict the coding regions and the sequence direction of unigenes that have no significant results in the search against protein databases above (Iseli and others 1999). The BLAST2GO program was applied to Gene Ontology analysis of unigenes using the best alignment results searched against the Nr database (Conesa and others 2005). Then the distribution of GO annotations was plotted by WEGO software (Livak and Schmittgen 2001). The KEGG and COG annotations were assigned using the searching result against KEGG and COG databases.
Unigene Expression Analysis
The Trinity toolkit was used to estimate the abundance of unigenes and indentify DEGs (Grabherr and others 2011). Reads of each library were mapped to the transcriptome assembly using bowtie with a maximum insert size of 800 (Langmead and others 2009). Using RSEM, the abundance of unigenes was estimated and assigned a FPKM value, and edgeR was used to identify DEGs (Mortazavi and others 2008; Pfaffl 2001; Robinson and others 2010). An FPKM filtering cut-off of 1.0 in at least one of the four libraries was used to determine expressed transcripts. When determining the DEGs, a cut-off of 1.0 in at least one the four libraries was used. Then DEGs were selected on the condition of p value ≤ 0.001 and |log2 FC| ≥ 1. GO functional enrichment analysis was performed using GO-TermFinder (Boyle and others 2004). KEGG pathway enrichment was performed using KOBAS 2.0 (Mao and others 2005).
Quantitative Real-Time PCR (qRT-PCR) of RNA-Seq Data
The DEGs involved in flowering were selected for confirmation by qRT-PCR analysis. Total RNA was extracted from the shoot tip tissues and surrounding core leaves and used for cDNA synthesis with the same procedures as described above. Real-time PCR reactions were performed on buffer (10 µl) composed of 0.2 µM of gene-specific primers (Supplementary Data 1), 50 ng of cDNA samples and 5 µl of 2 × SYBR Green Master Mix Reagent (Applied Biosystems). The gene for actin was used as an internal control to estimate the relative expression level of the analyzed genes. The thermal cycles were set as follows: 95 °C for 10 min, and 45 cycles of 95 °C for 5 s, 60 °C for 30 s. The relative expression of genes was calculated using the delta–delta Ct method of the system (Livak and Schmittgen 2001). The corresponding qRT-PCR efficiencies were calculated according to the mathematical model of Pfaffl (Pfaffl 2001). The qRT-PCR products were validated by both sequencing and gel electrophoresis. The qRT-PCR reactions were set up with three biological replications and three technical replicates per experiment.
Results
Illumina Sequencing and De novo Assembly
In this study, four cDNA libraries from adult and juvenile plants under ethylene treatment and water treatment were prepared and subjected to Illumina deep sequencing (Fig. 1). The RNA integrity numbers (RIN) of the four libraries were determined by RNA samples bioanalyzer firstly, which generated the RIN scores of 8.3, 7.1, 7.6, and 6.9 for the four samples, respectively. The outputs of sequenced data from adult plants with water treatment (A0), juvenile plants with water treatment (J0), adult plants with ethylene treatment (A1), and juvenile plants with ethylene treatment (J1) were 55,238,936, 53,797,292, 53,471,812, and 53,485,862 qualified Illumina clean reads, respectively, with 90 bp mean length. These reads of the four libraries were assembled into unigene sequences using de novo assembly software trinity (Grabherr and others 2011). Then, Unigenes of the four libraries were merged into a unified library with 86,609 sequences and a mean size of 987 bp, which included all non-redundant unigene sequences of the four libraries (Table 1). Figure 2 shows the distribution of transcript lengths, which range from 200 to 11,534.

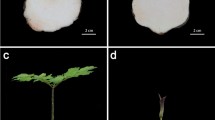
A. fasciata juvenile and adult plants. a Adult plants (right) and juvenile plants (left), b adult plants (right) and juvenile plants (left) after ethylene treatment 40 days

The length distribution of assembled sequences. The reads from four libraries were assembled into 86,609 transcripts
The CEGMA approach was used to assess the completeness of the transcriptome assembly, using a similarity search of the assembly with a set of 248 conserved eukaryotic core genes (CEGs) (Parra and others 2007). Of the 248 CEGs, 236 (95.16 %) genes are completely assembled and 247 (99.60 %) were partially assembled, indicating that the transcriptome assembly was of good completeness.
Functional Annotation
For annotation, unigene sequences of A. fasciata were first searched against the non-redundant (Nr) database of NCBI using BlastX with a cut-off e value of 1e-5. Of the searching results, 49,423 genes (57.06 % of unigene sequences) showed significant similarity to the proteins in the Nr database. The similarity distribution of the best matches is shown in Fig. 3a, 15.4 % of the matches were of high similarity ranging from 85 to 100, and 39.2 % of the hits were of similarity ranging from 60 to 80 %. Moreover, the species-based distribution of best matches is shown in the Fig. 3b. The result of homology analysis revealed that 18.47 % of the genes of A. fasciata showed the greatest similarity to Oryza sativa Japonica Group, while Vitis vinifera (13.99 %), Sorghum bicolor (9.91 %), Brachypodium distachyon (9.90 %), Zea mays (9.09 %), Oryza sativa Indica Group (6.00 %), and Hordeum vulgare subsp. Vulgare (3.82 %) showed a lower similarity to genes of A. fasciata. Then, these unigene sequences were searched against the Swiss-Prot database using a cut-off e value of 1e-5, with 35,251 genes (40.70 % of unigene sequences) returning an above cut-off BLAST result.

Summary for results of BlastX against NCBI NR database. a Similarity distribution of BlastX results. b Species-based distributions of BlastX results
GO annotations were used to classify the functions of the unigene sequences based on the result of BlastX searching against the Nr database using blast2go (Conesa and others 2005). Among the unigenes with significant hits in searching against the Nr database, 28,289 unigenes were categorized into 64 functional groups (Supplementary Data 2). Among the sub-categories of three main GO categories, ‘cell’ (10.6 %), ‘cell part’ (10.6 %), ‘organelle’ (8.6 %), ‘cellular process’ (7.9 %), ‘metabolic process’ (7.7 %), ‘catalytic activity’ (7.0 %), ‘binding’ (7.0 %) were the most represented categories. Only a few unigenes were classified into the categories of ‘virion,’ ‘viron part,’ ‘extracellular matrix part,’ ‘metallochaperone activity,’ ‘channel regulator activity,’ ‘protein tag,’ and ‘viral reproduction’ (Fig. 4).

GO assignments for transcriptome of A. fasciata
To further evaluate the completeness of our transcriptome library, we searched the annotated sequences against Clusters of Orthologous Group (COG) databases. In total, 19,293 sequences were assigned a COG classification (Fig. 5). These sequences were classified into 25 categories, of which the categories ‘General function prediction only’ (11.7 %), ‘Translation,’ ‘ribosomal structure and biogenesis’ (11.2 %), ‘Transcription’ (9.5 %), ‘Replication, recombination and repair’ (8.4 %), ‘Function unknown’ (8.3 %) were the top 5 categories (Supplementary Data 3). Moreover, of the 25 categories, ‘Defense mechanisms’ (13; 0.023 %) and ‘Nuclear structure’ (2; 0.0036 %) were the least represented.

COG Functional classification of transcriptome of A. fasciata
To identify the biological pathways that are active in A. fasciata, we mapped the unigene sequences to the reference canonical pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG). In total, we assigned 28,350 sequences to 129 KEGG pathways (Supplementary Data 4). Among the KEGG pathways, ‘Metabolic pathways’ (16.5 %), ‘Spliceosome’ (7.8 %), ‘Biosynthesis of secondary metabolites’ (5.4 %), ‘Endocytosis’ (4.4 %), ‘RNA transport’ (4.3 %), ‘Ether lipid metabolism’ (3.9 %), ‘Plant-pathogen interaction’ (3.6 %), and ‘mRNA surveillance pathway’ (3.2 %) were the highly represented pathways.
After a similarity search against transcription factor (TF) sequences in PlantTFDB using BlastX (e value < 1e-5), we identified 3347 putatively TFs (Jin and others 2013). These putatively TFs were assigned to 56 families according to the classification of TFs in Planttfdb (Supplementary Data 5). Among the TF gene families, bHLH (298) has the greatest number of transcripts, followed by C2H2 (233), MYB-related (215) and C3H (209). LFY (2), VOZ(3), and HRT-like (3) have fewer transcripts.
For the unigenes having no significant hits in searching against the protein databases above, the CDS of the unigenes are predicted by ESTScan (Iseli and others 1999) and then translated into peptide sequences. In the end, we obtained a “Blast-CDS” library with 49,538 unigene sequences and an ‘ESTscan-CDS’ library with 1499 EST sequences.
Unigene Expression Analysis
Expression analysis of four libraries was carried out using Trinity software. Clean reads from each library were mapped into merged transcriptome libraries. Then, the expression abundance of each gene was calculated and assigned a FPKM value. A total of 7748 DEGs were detected from the comparison of four libraries, of which 1231, 1328, 4879, and 3982 DEGs were identified from ‘A0_vs_J0,’ ‘A1_vs_J1,’ ‘A0_vs_A1,’ and ‘J0_vs_J1,’ respectively. The expression types of four libraries are shown in Fig. 6. As shown in Fig. 7, there were 671 upregulated genes and 651 downregulated genes for ‘A0_vs_J0,’ 787 upregulated genes and 444 downregulated genes for ‘A1_vs_J1,’ 1981 upregulated genes and 2898 downregulated for ‘A0_vs_A1,’ and 2084 upregulates genes and 1898 downregulated genes for ‘J0_vs_J1,’ respectively. We then investigated the distribution of DEGs in the TF gene families (Supplementary Data 5). Totally, 48 TF gene families had the number of DGEs ranging from 1 to 40. There were 13, 4, 23, 1, and 3 DEGs assigned into the TF gene families MIKC, CO-like, ERF, EIL, and AP2, respectively, which maybe involved in the regulation of flowering.

Log-fold changes in gene expression. a Log-fold changes in gene expression in ‘J1_vs_A1’. b Log-fold changes in gene expression in ‘J0_vs_ A0’. c Log-fold changes in gene expression in ‘J0_vs_A0’. d Log-fold changes in gene expression in ‘A0_vs_A1’

Differently expressed genes between A. fasciata juvenile and adult plants. ‘J1_vs_A1’ refers to the comparison between the juvenile and adult plant with ethylene treatment; ‘J1_vs_A0’ refers to the comparison between the juvenile and adult plant with water treatment; ‘J0_vs_J1’ refers to the comparison between the juvenile plant with water and ethylene treatment; ‘A0_vs_A1’ refers to the comparison between the adult plant with water and ethylene treatment; “P value < 0.001, the absolute value of Log2 fold change (Log2 FC) ≥ 2 and FPKM ≥ 1” were used as the threshold to determine the significance of gene expression differences
To further detect the function of DEGs, we performed a GO enrichment analysis based on the result of the search against the Nr database by the hypergeometric test (Supplementary Data 6). DEGs were enriched into 41 GO terms of which 27 terms including ‘binding,’ ‘catalytic activity,’ ‘organelle,’ ‘cell part’ were both enriched for the four DEGs sets. Additionally, 3 GO terms including ‘electron carrier activity,’ ‘transporter activity,’ and ‘structural molecule activity’ were enriched for DEGs of ‘J1_vs_A1’, while DEGs from ‘A0_vs_A1’ and ‘J0_vs_J1’ were both enriched into the categories of ‘electron carrier activity,’ ‘membrane-enclosed lumen,’ ‘cell junction,’ ‘symplast,’ ‘transporter activity,’ and ‘growth.’
To detect the pathways that DEGs were involved in, we carried out KEGG enrichment analysis. After the DEG sequences were searched against the KEGG database, DEGs were assigned into KEGG pathways. And the KEGG pathways were then assigned corrected p-values through the hypergeometric test. A total of 57 KEGG pathways were represented, of which 5 pathways including ‘Metabolic pathways,’ ‘Biosynthesis of secondary metabolites,’ ‘ABC transporters,’ ‘Plant-pathogen interaction,’ and ‘Spliceosome’ were enriched for the four DEGs sets. Comparing to DEGs in ‘J0_vs_A0,’ DEGs in ‘J1_vs_A1’ were enriched in ‘Phenylpropanoid biosynthesis,’ ‘Plant hormone signal transduction,’ ‘Other glycan degradation,’ ‘Isoquinoline alkaloid biosynthesis,’ ‘Phenylalanine, tyrosine and tryptophan biosynthesis,’ ‘Starch and sucrose metabolism,’ ‘Valine, leucine and isoleucine biosynthesis,’ ‘Cutin, suberine and wax biosynthesis,’ and ‘Carotenoid biosynthesis,’ while DEGs in ‘J0_vs_A0’ were not represented. Meanwhile, DEGs in ‘A1_vs_J1’ and ‘A0_vs_A1’ were also both enriched into ‘Glycerophospholipid metabolism,’ ‘Endocytosis,’ ‘Ether lipid metabolism,’ ‘NA transport,’ and so on; totally 10 pathways except the 5 pathways that the four DEGs set were both represented in, while a total of 24 pathways including ‘Pyrimidine metabolism,’ ‘RNA polymerase,’ ‘Phenylpropanoid biosynthesis,’ ‘Glycolysis/Gluconeogenesis,’ etc., that DEGs in ‘A1_vs_J1’ were enriched into but DEGs in ‘A0_vs_A1’ were not (Supplementary Data 7).
DEGs Involved in Flowering
We indentified 62 DEGs that may be involved in flowering from our transcriptome data (Table 2, Supplementary Data 8). These DEGs included several pathway genes that control flowering time and flowering development: (1) ethylene signaling pathway elements EIN4, CTR1, Ethylene-overproduction protein 1 (ETO1), and EIL3; (2) photoperiodic pathway and circadian clock-related genes, CO, GI, CRY2, LATE ELONGATED HYPOCOTYL (LHY), COP1, and HAP3; (3) AP2/ERF family genes, AP2-like ethylene-responsive TF At2g41710, TOE1, TOE2, TOE3, ERF Win1, ERF 1, ERF 109, and ERF 014; MADS-box family genes, MADS-box 14, MADS-box 23, and MADS-box 5; (4) E3 ubiquitin ligase-related genes, COP1, Cullin-1, and F-box family proteins; (5) PHYTOCHROME-INTERACTING FACTORs, PIF3 and PIF4; (6) GA pathway genes, GA2ox, GA20ox, GID1, and DELLA; (7) floral organ and meristem identity genes, APETALA 1 (AP1) and AGAMOUS (AG); (8) other genes involve in flowering timing and development, alpha, alpha-trehalose-phosphate synthase (TPS6, TPS7, TPS9), FD, CLAVATA 1 (CLV1), BEL1, cytochrome P450 90B1, cytochrome P450 734A6, serine/threonine-protein kinase BRI1, and ULTRAPETALA 1 (UTL1). According to the expression patterns of these DEGs, we classified these DEGs into three classes: (1) DEGs expressed differently in ‘J0_vs_A0,’ including COL3, PIN2, TOE3, CRY2, and so on; (2) DEGs expressed differently in ‘J1_vs_A1,’ including FT, TPS6, HAP3, GA2OX8, CTR1, CRY2, and so on; (3) DEGs expressed differently in ‘A0_vs_A1’ or ‘J0_vs_J1’ but not in ‘J0_vs_A0’ and ‘A1_vs_A0,’ including, WIN1, EIN4, ACC synthase 1, GID1, PIF3, BRI1, etc. Theoretically, the first class of DEGs may be involved in development of A. fasciata; the second class of DEGs may play key roles in flowering induction by ethylene in adult A. fasciata plants. The third class of DEGs may participate in the basic response of ethylene.
Of the DEGs involving flowering, 13 DEGs were selected to qRT-PCR analysis (Fig. 8, Supplementary Data 9). The expression of selected DEGs was examined in adult and juvenile plants under water and ethylene treatment, respectively. c2000030287_g1_i1 (FT), c100007569_g2_i1 (GI), c100001514_g3_i1 (AP1), and c2000048950_g1_i1 (MADS5) both increased in adult and juvenile plants under ethylene treatment compared to plants with water treatment, but increased more markedly in adult plants with ethylene treatment. c2000016847_g1_i1 (FD) and c100001523_g5_i1 (AP2) decreased both in adult and juvenile plants under ethylene treatment. The expression of these DEGs was analyzed by qRT-PCR corresponding with expression level in comparative transcriptome analysis, suggesting that the transcriptome profiles accurately reflected global transcriptome changes in response to ethylene treatment.

qRT-PCR validation of selected differently expressed genes. The amount of transcript was normalized to the level of A. fasciata actin like gene (homolog with Ananas comosus action gene gi|397881472). Mean values and standard errors (bars) were obtained from three biological replicates and three technical replicates
Discussion
We have performed comparative transcriptome analysis of two libraries from adult and juvenile plants under ethylene treatment in our previous study, with 52,560,902 and 52,396,972 Illumina clean reads, respectively. In this study, we constructed four transcriptome libraries of A. fasciata adult and juvenile plants under water and ethylene treatment, and achieved totally 86,609 sequences with a mean size of 987 bp, whereas we obtained a library of 71,445 unique sequences with a mean length of 461 bp in our previous study. Using BlastX to search against the Nr database with a cut-off E-value of 1e-5, 49,423 genes (57.06 % of unigene sequences) resulted in a significant hit; however, 35,483 genes (49.7 % of the unigene sequences) were matched to the Nr database in the previous study. With the more clean reads, we constructed a more complete and accurate transcriptome library, which will fascinate further analysis. Moreover, we obtained 10,036 DEGs between adult and juvenile plants under ethylene treatment in our previous study, which is more than the 1328 DEGs in this study, indicating that we reduced false positive predictions markedly in our DEG analysis.
ACC synthase is a key enzyme in ethylene synthesis and a key contributor toward flowering in mature pineapple (Trusov and Botella 2006). ACC synthase was upregulated in both ‘A0_vs_A1’ and ‘J0_vs_J1,’ implying that both adult and juvenile plants responded to ethylene by a burst of endogenous ethylene synthesis (Trusov and Botella 2006). FT protein is the major component of the mobile flower-promoting signal florigen, promoting the transition from vegetative growth to flowering in plants. Our data implying that the abundance of FT increased both in adult and juvenile plants in response to ethylene and dramatically increased in adult plants. After expression in leaves, FT proteins were transferred to the shoot apex and interacted with FD, resulting in increased expression of AP1 in the shoot apex and inducing subsequent flowering. FD is a TF required for transition to flowering promoted by FT (Abe and others 2005). FD is already expressed before floral induction in vegetative growing tissue and preferentially expressed in the shoot apex. FD mRNA was observed in leaf and floral anlagen, and was reduced soon after AP1 was expressed (Wigge and others 2005). The decrease of FD in our transcriptome data both in adult and juvenile plants may be associated with increasing abundance of FT protein in the shoot apex. The increased AP1 expression level and decreased abundance of FD mRNA in our transcriptome data suggested that the increased FT expression in leaves resulted in flowering induction in A. fasciata adult plants with ethylene treatment.
The photoperiod pathway controls the response to seasonal day length and acts in the leaves through a signaling cascade involving GI and the transcriptional regulator CO. The circadian clock controls the floral transition in many species. CIRCADIAN CLOCK ASSOCIATED1 (CCA1), LHY, and TIMING OF CAB EXPRESSION1 (TOC1) act as crucial players in regulation of the circadian clock. LHY is a negative regulator of TOC1 and PSEUDO RESPONSE REGULATORS 1 (PRR1) (Pineiro and Jarillo 2013). TOC1 acts as a negative regulator of GI (Bolouri Moghaddam and den Ende 2013). The abundance of LHY mRNA was downregulated in ‘J0_vs_A0.’ Further, our data showed that the abundance of GI expression was significantly increased under ethylene treatment in A. fasciata. GI positively regulated the expression of CO. The blue light photoreceptor cryptochrome CRY2 is upregulated in ‘A1_vs_J1’ and ‘A0_vs_J0,’ which may positively regulate the stability of CO. So, the increasing expression level of key photoperiodic pathway genes GI and CRY2 may result in increasing abundance of CO protein and upregulate FT transcription. CO bound with HAP3 and HAP5 through the CCT motif promotes flowering by enhancing expression of FT and SUPPRESSOR OF CONSTANS1 (SOC1) (Cai and others 2007; Wenkel and others 2006). HAP3 was highly upregulated in ‘A0_vs_J0’ and ‘A0_vs_A1,’ which could be the partial reason of the increased expression of FT. In summary, our data imply that genes in the photoperiod pathway play a key role in flowering induction by ethylene in A. fasciata.
FT acts as an integrator, merging various environmental and developmental cues such as temperature, photoperiod, GA, and plant age. The AP2 family, acting as repressors of FT, is also downregulated in ‘A0_vs_J0,’ including TOE1 and TOE3. TOE1 and TOE3 are the targets of miR172 and repressed by miR172. miR172 reduces the abundance of its targets. Additionally, GI is involved in this photoperiodic miR172 induction (Jung and others 2007). The increased abundance of GI may result in the amount of miR172 increasing. TOE1 and TOE3 and miR172 were correlated with plant development stages (Song and others 2013). Therefore, the AP2 family may result in the different response of A. fasciata adult and juvenile plants to ethylene treatment.
Regulation of GA concentration is primarily via the biosynthesis enzymes GA20ox and GA3ox, and inactivating enzymes GA2ox. GA20ox was upregulated and GA20x downregulated in ‘A1_vs_J1,’ indicating that the bioactive GAs increased in adult plants with ethylene treatment. Bioactive GAs promote binding of the GID1 receptor to DELLA and initiate DELLA degradation (Mutasa-Gottgens and Hedden 2009). DELLA reduces the amount of miR172 and represses the activator of FT, including SPL3, SPL4, and SPL5. DELLA also negatively regulates PIF4 activity, which activates the expression of FT under high temperatures (de Lucas and others 2008). It is suggested that DELLA proteins mediate transcriptional activation of the GID1, GA20ox, and GA3ox genes and the repression of DELLA transcription (Middleton and others 2012). The decreasing concentration of DELLA protein may lead to decreased GID1 expression. GID1 was downregulated in both ‘A0_vs_A1’ and ‘J0_vs_J1,’ suggesting that both adult and juvenile plants increased the synthesis of bioactive GA, resulting in decreased DELLA protein abundance. Additionally, BRs and ABA influence flowering time by regulating FLC. ABA inhibits flower transition by ABI5, which was downregulated in our data (Wang and others 2013). The differential expression of cytochrome P450 90B1, cytochrome P450 734A6, BRI1, and ABI5 in our transcriptome data indicated that ABA and BRs were also influenced by ethylene in the process of flowering induction by ethylene.
Trehalose-6-phosphate has been proposed to function as a proxy for carbohydrate status of plants and plays a key role in the presence of excess sugar (Matsoukas 2014). Plants integrate diverse environmental and endogenous signals to ensure the timely transition from vegetative growth to flowering (Wahl and others 2013). Tre6P is required for FT and TSF expression under an inductive photoperiod. TPS7 and TPS9 were upregulated in ‘A0_vs_A1’ and ‘J0_vs_J1,’ suggesting the abundance of Tre6p increased in response to ethylene. Recent studies suggested that ethylene and sugar may interplay by the GID1 and PIF family (Matsoukas 2014). Hence, the interplay between sugar and ethylene may influence flowering induction in A. fasciata.
It is proposed that AGAMOUS (AG) is involved in the control of organ identity during early development of flowers and acts as a C class cadastral protein through repressing A class floral homeotic genes such as APETALA1. TOE3 binds to the second intron of the AG gene (Jung and others 2014). Clavata1 (CLV1) and BEL1 act with CLV3 in a signal transduction pathway coordinating growth between adjacent meristematic regions and control the balance between meristem cell proliferation and differentiation. BEL1 also acts with SHOOT MERISTEMLESS, formatting the BEL1–STM complex to maintain the indeterminacy of the inflorescence meristem (Bellaoui and others 2001). The differential expression of AG, APETELA1, CLV1, and BEL1 implied that the arrival of FT in the shoot apex meristem initiated the transition from vegetative to reproductive development in A. fasciata.
It has been revealed that ethylene signaling interacts with the GA pathway by reducing endogenous levels of GAs (Davis 2009). Ethylene causes stabilization of EIN3 and EIN3-like proteins by inhibiting the activity of their protease SCF (Binder and others 2007). But our data indicated that bioactive GAs were increased under ethylene treatment. So, how GA and ethylene interplay in A. fasciata remains unclear and worth studying. Moreover, the mechanism of the AP2 family response to ethylene in A. fasciata needs further study.
Conclusions
This study provided a global survey of changes in transcriptomes of A. fasciata in response to ethylene. The analyses of transcriptome profiles imply that FT is upregulated markedly in the adult plant and resulted in flowering. The AP2 family genes, such as TOE1 and TOE3, may result in age-dependent flowering induction by ethylene. Moreover, the differential expressions of GI, DELLA, GAD1, and so on indicate that a complicated network participated in the induction of flowering by ethylene, which will help in future studies.
References
Abe M, Kobayashi Y, Yamamoto S, Daimon Y, Yamaguchi A, Ikeda Y et al (2005) FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309:1052–1056
Achard P, Baghour M, Chapple A, Hedden P, Van Der Straeten D, Genschik P et al (2007) The plant stress hormone ethylene controls floral transition via DELLA-dependent regulation of floral meristem-identity genes. Proc Natl Acad Sci USA 104:6484–6489
Andres F, Coupland G (2012) The genetic basis of flowering responses to seasonal cues. Nat Rev Genet 13:627–639
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 15:2730–2741
Bellaoui M, Pidkowich MS, Samach A, Kushalappa K, Kohalmi SE, Modrusan Z et al (2001) The Arabidopsis BELL1 and KNOX TALE homeodomain proteins interact through a domain conserved between plants and animals. Plant cell 13:2455–2470
Binder BM, Walker JM, Gagne JM, Emborg TJ, Hemmann G, Bleecker AB et al (2007) The Arabidopsis EIN3 binding F-Box proteins EBF1 and EBF2 have distinct but overlapping roles in ethylene signaling. Plant Cell 19:509–523
Bolouri Moghaddam MR, den Ende WV (2013) Sugars, the clock and transition to flowering. Fron Plant Sci 4:22. doi:10.3389/fpls.2013.00022
Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM et al (2004) GO:TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710–3715
Cai X, Ballif J, Endo S, Davis E, Liang M, Chen D et al (2007) A putative CCAAT-binding transcription factor is a regulator of flowering timing in Arabidopsis. Plant Physiol 145:98–105
Chuck G, Hake S (2005) Regulation of developmental transitions. Curr Opin Plant Biol 8:67–70
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Davis SJ (2009) Integrating hormones into the floral-transition pathway of Arabidopsis thaliana. Plant Cell Environ 32:1201–1210
de Lucas M, Daviere JM, Rodriguez-Falcon M, Pontin M, Iglesias-Pedraz JM, Lorrain S et al (2008) A molecular framework for light and gibberellin control of cell elongation. Nature 451:480–484
Fornara F, de Montaigu A, Coupland G (2010) SnapShot: control of flowering in Arabidopsis. Cell 141:550 e551–550 e552
Galvao VC, Horrer D, Kuttner F, Schmid M (2012) Spatial control of flowering by DELLA proteins in Arabidopsis thaliana. Development 139:4072–4082
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hua J, Meyerowitz EM (1998) Ethylene responses are negatively regulated by a receptor gene family in Arabidopsis thaliana. Cell 94:261–271
Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting evaluating and reconstructing potential coding regions in EST sequences. ISMB 99:138–148
Jung JH, Seo YH, Seo PJ, Reyes JL, Yun J, Chua NH et al (2007) The GIGANTEA-regulated microRNA172 mediates photoperiodic flowering independent of CONSTANS in Arabidopsis. Plant Cell 19:2736–2748
Jung CH, Wong CE, Singh MB, Bhalla PL (2012) Comparative genomic analysis of soybean flowering genes. PLoS One 7:e38250
Jung JH, Lee S, Yun J, Lee M, Park CM (2014) The miR172 target TOE3 represses AGAMOUS expression during Arabidopsis floral patterning. Plant Sci 215–216:29–38
Kieber JJ, Rothenberg M, Roman G, Feldmann KA, Ecker JR (1993) CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the raf family of protein kinases. Cell 72:427–441
Kumar SV, Lucyshyn D, Jaeger KE, Alos E, Alvey E, Harberd NP et al (2012) Transcription factor PIF4 controls the thermosensory activation of flowering. Nature 484:242–245
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Lingam S, Mohrbacher J, Brumbarova T, Potuschak T, Fink-Straube C, Blondet E et al (2011) Interaction between the bHLH transcription factor FIT and ETHYLENE INSENSITIVE3/ETHYLENE INSENSITIVE3-LIKE1 reveals molecular linkage between the regulation of iron acquisition and ethylene signaling in Arabidopsis. Plant Cell 23:1815–1829
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 − ΔΔCT method. Methods 25:402–408
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21:3787–3793
Matsoukas IG (2014) Interplay between sugar and hormone signaling pathways modulate floral signal transduction. Frontiers in genetics 5:218
Merchante C, Alonso JM, Stepanova AN (2013) Ethylene signaling: simple ligand, complex regulation. Curr Opin Plant Biol 16:554–560
Middleton AM, Ubeda-Tomas S, Griffiths J, Holman T, Hedden P, Thomas SG et al (2012) Mathematical modeling elucidates the role of transcriptional feedback in gibberellin signaling. Proc Natl Acad Sci USA 109:7571–7576
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Mutasa-Gottgens E, Hedden P (2009) Gibberellin as a factor in floral regulatory networks. J Exp Bot 60:1979–1989
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res 29:e45
Pineiro M, Jarillo JA (2013) Ubiquitination in the control of photoperiodic flowering. Plant science : an international journal of experimental plant biology 198:98–109
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Sawa M, Kay SA (2011) GIGANTEA directly activates Flowering Locus T in Arabidopsis thaliana. Proc Natl Acad Sci USA 108:11698–11703
Schaller GE (2012) Ethylene and the regulation of plant development. BMC Biol 10:9
Solano R, Stepanova A, Chao Q, Ecker JR (1998) Nuclear events in ethylene signaling: a transcriptional cascade mediated by ETHYLENE-INSENSITIVE3 and ETHYLENE-RESPONSE-FACTOR1. Genes Dev 12:3703–3714
Song YH, Ito S, Imaizumi T (2013) Flowering time regulation: photoperiod- and temperature-sensing in leaves. Trends Plant Sci 18:575–583
Trusov Y, Botella JR (2006) Silencing of the ACC synthase gene ACACS2 causes delayed flowering in pineapple [Ananas comosus (L.) Merr.]. J Exp Bot 57:3953–3960
Wahl V, Ponnu J, Schlereth A, Arrivault S, Langenecker T, Franke A et al (2013) Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 339:704–707
Wang Y, Li L, Ye T, Lu Y, Chen X, Wu Y (2013) The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J Exp Bot 64:675–684
Wenkel S, Turck F, Singer K, Gissot L, Le Gourrierec J, Samach A et al (2006) CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis. Plant Cell 18:2971–2984
Wigge PA, Kim MC, Jaeger KE, Busch W, Schmid M, Lohmann JU et al (2005) Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309:1056–1059
Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS (2009) The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138:750–759
Yu S, Galvao VC, Zhang YC, Horrer D, Zhang TQ, Hao YH et al (2012) Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA promoter binding-like transcription factors. Plant Cell 24:3320–3332
Zhong S, Zhao M, Shi T, Shi H, An F, Zhao Q et al (2009) EIN3/EIL1 cooperate with PIF1 to prevent photo-oxidation and to promote greening of Arabidopsis seedlings. Proc Natl Acad Sci USA 106:21431–21436
Zuo Z, Liu H, Liu B, Liu X, Lin C (2011) Blue light-dependent interaction of CRY2 with SPA1 regulates COP1 activity and floral initiation in Arabidopsis. Curr Biol 21:841–847
Acknowledgments
The research was supported by the The National Natural Science Foundation of China (31372106) and The Fundamental Scientific Research Funds for CATAS-TCGRI (1630032014018).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhiying Li and Jiabin Wang have been contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Z., Wang, J., Zhang, X. et al. Transcriptome Sequencing Determined Flowering Pathway Genes in Aechmea fasciata Treated with Ethylene. J Plant Growth Regul 35, 316–329 (2016). https://doi.org/10.1007/s00344-015-9535-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-015-9535-4