
Abstract
TiO2 samples with two different morphology were synthesized via sol–gel and hydrothermal techniques. The effect of synthesis procedure on structural, morphological, optical, and electrical properties was studied. Rietveld analysis revealed that anatase phase having tetragonal structure dominates at room temperature. The presence of anatse phase of TiO2 was further confirmed by the analysis of various peaks obtained from the Raman spectra. The field-emission scanning electron microscopy (FESEM) and transmission electron microscopy micrographs depicted the formation of two different kinds of morphologies with average particle size ranging from 9.87 to 11.35 nm. The FESEM micrographs showed homogeneous particle distribution for sol–gel synthesized sample, whereas it depicted rod-like structure in the sample synthesized via hydrothermal technique. The X-ray photoelectron spectroscopy analysis clearly indicated the presence of appropriate chemical composition and valency states of Ti and O element in TiO2 samples. The optical bandgap was estimated from the UV–visible spectra and found to be in corroboration with the reported values. The conductivity spectra were analyzed using Jonscher power law. The values of activation energy suggested that the conduction mechanism is thermally activated. The conductivity isotherms were scaled through Ghosh scaling model. In the sol–gel synthesized sample, the conduction mechanism was found to be independent of temperature in the entire measured temperature range; however, the hydrothermally synthesized sample depicted that the conduction mechanism is temperature-dependent in the measured temperature range. This discrepancy was understood in terms of structural changes and charge trapping within the structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In previous years, numerous reports have been published on experimental and theoretical studies of titanium dioxide (TiO2) semiconductor material [1, 2]. The nanosized TiO2 has received an immense interest due to a large number of applications in photocatalytic systems, sensors, water splitting, medical implants, fuel cells, and solar cells, etc. [3,4,5,6]. It is one of the extensively studied semiconductor materials utilized in many applications due to its commercial availability at low cost, chemical stability, nontoxic nature, and general reactivity [7]. Among other oxide materials such as tungsten oxide (WO3), tin oxide (SnO2), zinc oxide (ZnO), etc., the TiO2 achieve remarkable interest. TiO2 has three distinct polymorphs at room temperature and pressure: brookite, rutile, and anatase. The anatase and rutile phases of TiO2 consist of the tetragonal structure while brookite having an orthorhombic crystal structure. The bandgap corresponds to anatase, rutile, and brookite are 3.2 eV, 3.0 eV, and 3.3 eV, respectively [7]. In bulk form, rutile and in nanorange anatase are more stable than that of the other polymorphs [8]. Anatase phase has an appropriate crystal structure for rapid electron mobility and wide bandgap in comparative to rutile for photocatalysis and solar-electric conversion on efficiency [9]. Several methods such as sol–gel, sputtering, chemical vapor deposition, physical vapor deposition, and hydrothermal, have been used for the synthesis of nanorange TiO2 [3, 7, 10,11,12,13,14,15,16].
Experimentally, it is established that the processing conditions influence the microstructural and electronic properties of TiO2 [7]. Tripathi et al. reported the effect of sol–gel and co-precipitation method on photoconducting and optical properties of TiO2 system [17]. Takeda et al. explained the improved photocatalytic activity of TiO2, synthesized by the DC magnetron sputtering [14]. Dar et al. reported the controlled synthesis of TiO2 nanospheres and nanoparticles through a microwave-assisted technique for the application of photoanode in dye-sensitized solar cells [9]. Yuvapragasam et al. describe that the TiO2 nanorods can be synthesized by hydrothermal method, arranged in a broccoli-like manner having the potential application in the field of photovoltaics [18]. The influence on electrical properties due to the microstructural feature was studied by Song et al., and they found the columnar shape kind of morphology and described the ionic conduction dominating feature in electrical conductivity of TiO2 [19]. Mardare et al. investigated the physical properties of TiO2 thin films synthesized by d.c. sputtering, and represented that the deposition condition highly influences the structural, morphological, optical, and electrical properties [20]. Furthermore, the electrical conductivity of TiO2 at high temperature (T > 300 K) can be explained by thermally activated conduction mechanism [20, 21], and at low temperatures, it is described by hopping via impurity centers [12].
In the last couple of decades, the phenomenon of ionic and electronic conduction in disordered solids such as polymers, glass, and semiconductors are highly fascinating the researchers. Among all the methods used to describe the charge carrier dynamics of these materials, the conductivity spectroscopy is most commonly used [22]. The variation of the real part of frequency-dependent conductivity can be well defined using Jonscher’s power law [23]. Furthermore, the temperature and frequency-dependent conductivity also follow the time–temperature superposition principle (TTSP). This suggests that all the conductivity isotherms merge into a single master curve. Variety of scaling models proposed by multiple researchers such as Summerfield scaling [24], Rolling et al. scaling [22], Sidebottom scaling [25], and Ghosh scaling [26], etc. These models are widely following on glasses, perovskite, composite, and semiconductor systems.
Although, numerous research have been done on the variation of processing conditions, structural, morphological, optical, and electrical properties of the TiO2 system. However, there is no literature which is available on the dynamics of ions and scaling nature of conductivity spectra of this system. Hence, it appears essential to investigate the behavior of conductivity isotherms and scaling properties of the TiO2 nanopowders. The prime objective of this work is to investigate the influence of synthesis condition on the electrical and scaling properties of TiO2 samples. Therefore, the undoped TiO2 nanopowders were synthesized by two different synthesis methods: sol–gel and hydrothermal techniques. The effect of processing conditions on the morphology of TiO2 nanomaterial was investigated. Besides this, the ion dynamics of the material and scaling behavior of the conductivity and modulus spectra in a measured temperature range are discussed. In addition to it, the temperature and microstructure dependent conduction mechanisms have been proposed for the TiO2 samples.
2 Sample preparation and characterization
2.1 Synthesis of nanoparticles via sol–gel route
Titanium tetraisopropoxide (TTIP), isopropanol, nitric acid, and deionised water were used to synthesize the TiO2 nanoparticles by the sol–gel method through the hydrolysis of TTIP. The schematic diagram of the synthesis process is shown in Fig. 1. For the synthesis process, 5 ml of TTIP was dropped slowly into the vigorously stirred 6 ml isopropanol solution. The obtained mixture was stirred for 15 min, and furthermore, 75 ml of deionized water was added drop-wise into this mixture resulting in the formation of white precipitate. After a few seconds, this white precipitate gets converted into the white solution. Then, the solution was stirred thoroughly by controlling its temperature at 80 °C for 1 h. 2.5 ml of acetic acid was added drop-wise to this solution and the resulting solution was stirred for 5 h. After 5 h of stirring, 1 ml of nitric acid was added drop-wise for the peptization and the resulting solution was allowed to stir for 2 h. The as-formed solution was cooled naturally up to room temperature and dried at 60 °C in the vacuum oven. The obtained powder was ground in a mortar pestle to make a fine powder and then finally annealed at 500 °C for 3 h.

Schematic diagram of the synthesis of TiO2 nanoparticles by sol–gel method
2.2 Synthesis of nanorods via hydrothermal technique
TiO2 powders were also synthesized through the hydrothermal treatment of 2 g TiO2 powder in 50 ml of 10 M NaOH solution and stirred for 10 min at the room temperature. This solution was then transferred into a Teflon lined stainless steel autoclave and the condition for the hydrothermal reaction was maintained thoroughly at 150 °C for 48 h. Furthermore, the autoclave was cooled down naturally up to the room temperature and resulting in the formation of white salt cake type of mixture. The upper residual liquid was poured off. The obtained residual dispersion was washed several times with 0.1 M HCl and distilled water until the solution reached up to the pH of 7. The obtained sample was filtered and dried at 80 °C in a vacuum oven for 8 h. To obtain the fine powder, the sample was ground in a mortar pestle and annealed at 500 °C for 4 h. Figure 2 represents the schematic diagram of the above synthesis route.

Schematic diagram of the synthesis of TiO2 nanorods by hydrothermal technique
2.3 Characterization techniques
To determine the crystal structure and the phase purity, the samples were characterized by X-ray diffraction (XRD) technique using Rigaku smart lab X-ray diffractometer with CuKα radiation. The morphologies of the samples were investigated by the field-emission scanning electron microscopy (FESEM) (Oxford instrument SEM EVO 18), transmission electron microscopy (TEM) (FEI TECNAI G2 20 TWIN Type, FEI, USA), and selected area electron diffraction (SAED). The size distribution and the average diameter of particles were calculated by measuring the mean size of particles from TEM micrographs obtained from the different regions of grids. The compositional analysis was performed by energy-dispersive X-ray spectroscopy (EDX). The optical absorbance spectra were recorded using JASCO V-770 UV–visible spectrometer and bandgaps were calculated using the Tauc’s relation. The X-ray photoelectron spectroscopy (XPS) spectrum of samples was measured by KRATOS (Amicus model) high-performance analytical instrument using the Mg target under 10–6 Pa pressure. The Raman spectra of samples were detected using DXRxi Raman imaging microscope by Thermo SCIENTIFIC with the excitation wavelength of 532 nm, laser power = 6.1 mW, exposer time = 0.100 s, and the number of scan = 60. For the electrical measurement, the powder samples were pelletized in the cylindrical disk using a hydraulic pressing machine and silver paste was used as an electrode. Furthermore, the electrical measurement of silver-coated pellets was recorded in the air by the two-probe technique using LCR meter (Wayne Kerr 6500P) over the frequency of range 20 Hz–1 MHz and in the temperature range 200–300 °C. The two parameters conductance (G) and dielectric loss (D) were recorded with respect to frequency and temperature. The modulus parameters were calculated using the formula:
where w is angular frequency, w is the 2πf of an applied electric field, and f being the frequency in cycles/s. C0 is the geometrical capacitance.
3 Results and discussion
3.1 Structural studies
3.1.1 X-ray diffraction
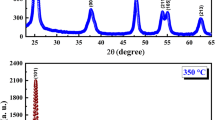
TiO2 nanoparticles, synthesized by sol–gel method, are referred hereafter as TP and rods like structure, obtained from hydrothermal technique, are indicated by TR. The nanoparticles and nanorods are further confirmed in our FESEM and TEM images and to be discussed in the forthcoming section. The crystal structure of TiO2 samples were first examined by X-ray diffraction technique and the patterns are shown in Fig. 3. The observed diffraction peaks were assigned as the TiO2 anatase phase and are well matched with JCPDS card (no. 21-1272), which shows that present syntheses process leads to the formation of single-phasic TiO2 nanostructures [27]. It confirms the phase formation of the tetragonal crystalline structure with space group I41/amd (#141) [28]. To obtain the information about structural parameters of synthesized samples, the Rietveld refinement of XRD patterns was performed using the FullProf software suite and the refined patterns are represented in Fig. 3. The refinement was executed by applying the tetragonal structure and space group I41/amd. The peak profiles were modeled with pseudo-voigt function. The values of fitting parameters Rwp (R-weighted pattern), Rp (R-pattern), Rf, and RBragg along with the structural parameters are reported in Table 1. The wider peaks in the diffraction pattern reveal the formation of nano structure of TiO2. It is important to note that the lattice parameters ‘a’ and ‘b’ are slightly higher in TP in comparison to TR. On the other hand, the slight increment has been observed in the value of lattice parameter ‘c’, tetragonal strain (c/a), and cell volume for TR.

Rietveld refinement of the X-ray diffraction pattern of TiO2 (TP and TR) systems
The crystallite size (D) and lattice strain (ɛ) affect the peak broadening, which can be induced by the structural defects. These two parameters can be calculated by the Williamson–Hall method using the following equation [29]:
where \(\beta_{hkl}\) is the peak width at half of the maximum intensity and k is a constant equal to 0.94. λ is the wavelength of the Cu-Kα radiation and \(\theta_{hkl}\) is the peak position. The term \(\beta_{hkl} \cos (\theta_{hkl} )\) was plotted against \(4{\sin}(\theta_{hkl}\)) showing a straight line [30]. From the fitted line, crystallite size and lattice strain can be determined using the y-intercept and slope, respectively. The values of crystallite size and lattice strain obtained from the W–H plot are summarized in Table 1. The crystallite size of the samples TP and TR is found to be 8.24 ± 0.8 nm and 10.44 ± 0.9 nm, respectively. The value of lattice strain in the case of sample TP is more than TR. While analyzing the XRD pattern carefully, it has been noticed that the peaks are slightly broad in sample TP in comparative to TR. This broadening in peaks arises due to the less crystallite size of TP in comparison to TR.
3.1.2 Morphology and microstructure analysis
The morphology of the powder samples was investigated by field-emission scanning electron microscopy. The micrograph for sample TP (Fig. 4a) reveals the formation of very fine particles, densely packed with spherical aggregation, while for sample TR (Fig. 4b), fine particles are aggregated in a rod-like network.

FESEM micrograph of sample a TP and b TR, TEM images of c TP and d TR, particle-size histogram of e TP and f TR, respectively
The detailed morphological and structural investigations were carried out through TEM. Morphologies observed by TEM bright-field images acquired for TP and TR samples are shown in Fig. 4c, d, respectively. The TP displays nearly spherical shaped particles with uniform size distribution having average size ~ 10 nm. Nanorods are observed in TR with an average width of ~ 11 nm. Size distribution histograms of TP and TR are presented in Fig. 4e, f, respectively, and evaluated using the lognormal function to obtain average particle size (D) and standard deviation (σ). The measured particle sizes fall in line with the XRD data. Figure 5a, b shows the selected area diffraction patterns corresponding to images shown as insets in TP and TR, respectively. Polycrystalline rings were indexed and phase was found to those of tetragonal anatase TiO2, further substantiate our XRD results. Representative high-resolution phase-contrast images acquired for TP and TR are presented in Fig. 5c, d, respectively. Lattice fringe spacing of ~ 0.34 nm corresponding to (101) interplanar distance of anatase TiO2 has been observed for TP (Fig. 5c). In addition to this, magnified image of a crystal oriented along [001] direction as inset along with FFT is also depicted. In the case of TR, HRTEM image shows lattice fringe spacing of ~ 0.34 nm (shown as an inset in Fig. 5d) which corresponds to (101) interplanar spacing of anatase TiO2. Moreover, an interesting observation pertaining to structural variants in TR sample has been seen, and representative HRTEM image is presented in Fig. 5e. Lattice fringe spacing of ~ 0.32 nm has been measured which corresponds to the interplanar distance of (110) and \((\bar{1}10)\) planes of tetragonal rutile phase of TiO2. However, the angle between the two planes is measured to be ~ 82° instead of 90°, which suggests that nanorod under present investigation is a variant of rutile TiO2. Aforesaid observation confirms that TR contains anatase as a major phase with the presence of trace amount of rutile also. This is further supplemented by XRD and TEM diffraction patterns discussed previously. The presence of rutile TiO2 as structural variants in TR might have far-reaching implications in its electrical transport behavior.

Selected area diffraction patterns with BF image as insets a, b and HRTEM images c, d of TP and TR, respectively. e High-resolution phase-contrast image and corresponding FFT pattern as an inset of rutile tetragonal phase in TR
3.1.3 Raman spectroscopy
To investigate the detailed structure of TP and TR samples, the Raman spectra were recorded, as shown in Fig. 6. The characteristic six Raman active vibrational modes (Eg(1), Eg(2), B1g, A1g + B1g, Eg(3)) corresponding to anatase phase of TiO2 tetragonal crystalline structure with space group I41/amd [31] for both of the samples prescribed by Bilbao crystallographic server, which was found to be in a good agreement with the XRD data. The high-intensity mode Eg(1) corresponds to the symmetric stretching vibration of oxygen atoms in O–Ti–O bond recorded at 148.37 cm−1 and 146.23 cm−1 for sample TP and TR, respectively. The B1g mode falls at 398.862 cm−1 and 396.9335 cm−, for TP and TR, respectively, which arises due to the symmetric bending vibrations of O–Ti–O bonds. Another mode A1g originates due to the antisymmetric vibration of O–Ti–O bonds. Raman modes at 520.3553 cm−1 in TP and 516.4984 cm−1 in TR belong to the overlapping of A1g and B1g modes that arise due to the vibration of O–Ti–O bond with the immovable Ti atoms [32]. The Raman spectrum of TP displays the shifting and broadening in the peaks corresponding to each and every vibrational mode as compare to TR shown in the inset of Fig. 6. This shift in frequencies and broadening can be attributed to a decrease in the crystallite size. The decrement in crystallite size influences the phonon modes and the electron–phonon interaction. As the crystallite size decreases, decrement in the interatomic distances takes place due to the contraction of volume and this may further lead to the enhancement in the value of force constant [13]. In the vibrational spectrum, the wavenumber (ν) is proportional to the square root of force constant. Therefore, increment in the force constant leads to the shifting of Raman modes towards the higher wavenumber side. Thus, the decrease in crystallite size may be attributed to the shifting to Raman modes in sample TP towards the higher wavenumber side [33]. Another reason for the shift in Eg(1) mode of O–Ti–O bond in the Raman spectra indicates the presence of oxygen vacancies in the sample [34]. Due to the existence of oxygen vacancies, lattice contraction takes place in TiO2. This indicates the shift in Raman peak towards higher wavenumber.

Room-temperature Raman spectra of TiO2 anatase TP and TR systems in wavenumber range 50–800 cm−1. Inset shows that the Raman spectra correspond to Eg(1) mode in the wavenumber range of 100–200 cm−1
3.1.4 XPS spectroscopy
To identify the oxidation states and composition of the constituent elements, X-ray photoelectron spectroscopy (XPS) technique was performed. Figure 7a shows the XPS survey spectrum of sample TP and TR. The wide range spectrum shows the presence of Ti, O, auger peaks, and a small fraction of adventitious carbon. The peak at ~ 284.8 eV attributes to the C 1s peak and arises due to the surface adsorbed CO2 or external contamination. The binding energies were determined after calibration with respect to the reference peak of C 1s. Figure 7b, c represents the Ti 2p spectrum, consisting of Ti 2p3/2 and Ti 2p1/2 signals that are positioned at binding energies of 458.6 eV and 464.4 eV, respectively [35]. The peak separation between Ti 2p3/2 and Ti 2p1/2 was evaluated as 5.8 eV which is in well agreement with the reported values [36]. This indicates the Ti4+ oxidation state of titanium in TiO2 matrix. Through the peak fitting, this spectrum may be divided into two contributions of Ti4+ (458.9 eV and 464.8 eV) and Ti3+ (458.4 eV and 463.9 eV) [37], as shown in Fig. 7b, c. The XPS result shows that the area of the peak corresponding to Ti3+ is more than the peak area of Ti4+ content in sample TP. On the other hand, in sample TR, the peak area corresponds to Ti4+ is higher than the peak area of Ti3+. This indicates that there are two types of the contribution of titanium species on the surface of TiO2 samples [35]. Furthermore, Fig. 7d, e shows the broader and asymmetric nature of oxygen (O 1s) peak and can be divided into two peaks at binding energy 530 eV and 531.9 eV. The peak situated at 530 eV refers to the lattice oxygen, while the peak at 531.9 eV indicates the presence of adsorbed oxygen [34]. On comparing the O 1s spectra of sample TP and TR, it has been noticed that the area of the peak corresponds to adsorbed or hydroxyl oxygen for sample TP is greater than that of TR. This also indicates the presence of Ti3+ defect sites on the surface of the samples. The higher value of the area for adsorbed oxygen suggests the formation of oxygen vacancies in the lattice for sample TP is more. This is in well agreement with our Raman measurement results.

XPS a survey spectrum, b, c core-level spectra of Ti 2p state, d, e O 1s state of sample TP and TR, respectively
3.2 Bandgap using UV–visible
The optical properties of TiO2 samples have been analyzed using UV–visible spectroscopy measurement. The room-temperature optical absorbance spectra of the samples have been investigated in the wavelength range of 200–800 nm and shown in Fig. 8. To determine the absorption edge wavelength, one can extrapolate the linear region of absorption spectra up to the x-axis. The absorbance edge for samples TP and TR is observed at 408 nm and 403 nm, respectively. It has been observed that the absorbance edge is shifted towards the higher wavelength side for sample TP. This shift indicates the decrease in the optical bandgap Eg.

a The UV–visible absorption spectra of TP and TR samples in a wavelength range of 200–800 nm. b Optical bandgap calculated from Tauc’s relation between (αhν)2 and photon energy (hν). Inset of b shows the images of TP and TR samples when they are exposed to daylight, UV-C light (shorter wavelength), and UV-A light (longer wavelength)
The optical energy bandgap of both the samples was calculated by analyzing the optical absorbance data for direct bandgap using Tauc’s relation [38]:
where A is the absorption edge width parameter, h is plank’s constant, α is the absorption coefficient. Eg is the optical direct band-gap energy of the material, and ν is the frequency of the incident photon. The optical bandgap Eg can be evaluated through the intercept by extrapolating the linear region [(hν) on the x-axis] of the Tauc plot where (αhν)2 = 0 [39]. The obtained values of bandgaps are indexed in Table 1, and it has been found that the direct optical bandgap for TP and TR is 3.19 eV and 3.25 eV, respectively, which is well consistent with the reported values in the literature [40, 41].
For further verification of these observations, the powder samples were exposed to daylight and UV lights [42]. Inset image of Fig. 8b shows the change in colors of powder samples when exposed to daylight, UV light with short-wavelength range (UV-C; 100–280 nm), and UV light with long-wavelength range (UV-A; 315–400 nm). While exposing in daylight, both samples TP and TR appear white in color, in UV-C light, it changes to pale yellow. When further exposed to UV-A light, both samples displayed the black color.
Further absorption along with transitions between states of band tails, arise due to structural defects, can be calculated through the absorption coefficient (α). The absorption coefficient has an exponential dependence on Urbach tail energy [42] and photon energy, represented by the relation α = α0 exp (hν/Eu), where Eu is the Urbach energy and α0 is the constant. The obtained values of Eu are 0.24 eV and 0.33 eV for sample TP and TR, respectively.
3.3 Electrical properties
The electrical properties of specimens TP and TR were explored by conductivity spectroscopy. The variation of the real part of frequency-dependent electrical conductivity for both samples at a few selected temperatures is shown in Fig. 9. The total conductivity in the conductivity spectrum is the combination of two contributions; one corresponds to low-frequency plateau [DC conductivity (σdc) and frequency-independent part] and another is dispersion region with increasing frequency (AC conductivity, σac, i.e., frequency-dependent part) [43]. The low-frequency region represents the dc conductivity and originates due to the random hopping motion of mobile charge carriers. The dispersive region corresponds to a combination of grain conductivity and grain boundary dielectric relaxation and emerges due to the forward–backward hopping of the charge carriers [44]. The conductivity for solids, polymers, glasses, crystal, nanocomposites, and semiconductors follows the Jonscher’s power law [23], which describe the frequency dependence of real part of the conductivity σ′(ν) given by the following equation [45]:
where ν is the frequency, \(\nu_{{\text{H}}}\) is the hopping frequency, and p is the power exponent usually smaller than unity and shows the electrical relaxation behavior of any material. The frequency \(\nu_{{\text{H}}}\) represents the onset frequency representing a change from dc to the dispersive conductivity region. The conductivity spectra represented in Fig. 9 can be described using the above equation. In the figure, the symbols represent the experimental data and the solid lines correspond to the fitted data points using Eq. (5). The fitting parameters, dc conductivity \(\sigma_{{{\text{dc}}}}\), hopping frequency \(\nu_{{\text{H}}}\), and power exponent p were obtained for both the samples at different temperatures.

Conductivity spectrum of sample a TP and b TR at various temperature range. Here, symbols represent the experimental data points and solid lines refer to fitted Jonscher equation
The activation energy of conduction for both samples was calculated by fitting the obtained conductivity data using Arrhenius relation [46]:
where Ea is the activation energy for conduction, σo is a pre-exponential factor, k is the Boltzmann constant, and T is the absolute temperature. Figure 10 shows the variation of log10(σdc·T) and 1000/T for sample TP and TR. For sample TP, only one slope is obtained in the entire measured temperature range and the value of activation energy (Ea) is obtained as 0.33 eV. However, two slopes are observed for sample TR in the measured temperature range of 200–300 °C and each slope corresponds to the different activation energy. In the temperature range 200–240 °C (region I), the activation energy for sample TR is 0.30 eV, and in the temperature range 240–300 °C (region II), its value is obtained as 0.52 eV. This suggests the presence of two different conduction mechanisms for TR in the measured temperature range. The change in the conduction mechanism in sample TR can also be understood in terms of scaling behavior. For this, Ghosh scaling [26] has been used.

Arrhenius representation for dc conductivity of sample TP and TR. Here, symbols show the experimental data and solid lines correspond to the linear fit to data points
The conductivity spectrum of various ion-conducting materials follows the time–temperature superposition principle (TTSP), i.e., the existence of single master curve for all conductivity spectra at different temperatures using reduced scaling parameters with respect to frequency and conductivity. This suggests that the spectral shape of conductivity spectra is independent of temperature and conduction mechanism does not depend on the temperature of the material. The mathematical expression of the principle can be given by the following [26]:
where scaling factor F is independent of temperature and \(\nu_{{\text{H}}}\) is temperature-dependent scaling frequency. To understand the insight of the conduction mechanism of the material, many models have been proposed by several researchers [24, 25, 47]. In the present work, Ghosh scaling is taken into account due to its wide applicability for many systems. In this scaling model, hopping frequency (νH) and dc conductivity (σdc) are taken as scaling parameters [48]. Figure 11 shows the scaled conductivity spectra of both the samples at different temperatures. From Fig. 11a, it can be observed that for composition TP, all the spectra merge into a single master curve over the entire measured temperature range. Thus, compound TP follows the time–temperature superposition principle (TTSP) in the given temperature range, suggesting that the conduction mechanism for TP is independent of temperature in the measured temperature range. From Fig. 11b and inset, for sample TR, the conductivity spectra does not collapse into a single master curve in the investigated temperature range of 200–300 °C, which can be expected as we did not get a single slope between log(σdc) and 1000/T (Fig. 10). Nevertheless, we find that the conductivity isotherms in temperature range from 200–240 and 240–300 °C superimposed nicely into a single master curve with minimal deviation, as shown in Fig. 11c, d. Hence, from Fig. 11, we can predict that the relaxation mechanism of sample TP in entire investigated temperature range is independent of temperature and the sample TR shows the temperature-independent behavior of relaxation mechanism only in temperature ranges of 200–240 °C and 240–300 °C but not in entire temperature range from 200–300 °C.

Scaled conductivity spectra of sample a TP and b–d TR, respectively
3.3.1 Modulus spectroscopy
For a closer investigation of the relaxation dynamics of the samples, the electric modulus is often used. It provides the response corresponding to bulk and excludes the electrode polarization effect [49]. The variation of the imaginary part of electric modulus M" with frequency at different temperatures ranging from 200–300 °C is shown in Fig. 12a, b. The low-frequency region of these peaks refers to long-range sub diffusive mechanism migration of charge carriers, i.e., mobile charge carriers can hop successfully through defect sites [50]. The high-frequency region of the peak attributes to short-range migration of the charge carriers, i.e., confinement of charge carriers into their potential wells [51]. With increasing frequency, the region where peak arises illustrates the transition from long- to short-range mobility [52]. From Fig. 12, on increasing temperature, the shift in modulus M″ peak towards higher frequency side is observed for both samples. As a consequence, the conductivity relaxation time τmax (= 1/νmax), obtained from the modulus relaxation frequency νmax corresponding to the maximum peak value of M″ (M″max), decreases as the temperature increases due to the fast movement of charge carriers [53]. The variation of conductivity relaxation time with temperature is plotted in Fig. 12c and described by the Arrhenius relation given by the equation \(\tau = \tau_{{\text{o}}} \exp (E_{{\text{a}}} /kT)\), where τ0 refers to the characteristic relaxation time. The value of τ0 can be obtained from the intercept of the linear fitted data between log τ and 1000/T plot. For sample TP, the value of τ0 reveals the fast relaxation time throughout the temperature range 200–300 °C i.e. order of 10.12 × 10–9 s, whereas for sample TR, its value varies from 19.44 × 10–6 s for temperature range 200–240 °C and 3.60 × 10–9 s for 240–300 °C. The relaxation time vs. 1000/T plot also showed a change in conduction mechanism for sample TR. Therefore, it can be concluded for sample TP and TR that the relaxation phenomenon is thermally activated. The nature of modulus indicates the existence of charge carrier hopping mechanism in the samples.

The variation of M″ with frequency for samples: a TP and b TR, respectively. c Arrhenius representation of conductivity relaxation time. Here, symbols show the experimental data and solid lines correspond to the linear fit to data points
To understand and verify the scaling behavior more precisely, scaling nature of modulus spectra of the samples was studied. Here, imaginary modulus axis has been scaled by the maximum value of M″ (i.e., M″max) and frequency axis is scaled by the maximum value of ν (i.e., νmax) [53, 54]. Figure 13 represents the normalized spectra of M″/M″max versus log(ν/νmax) in the temperature range of 200–300 °C for sample TP and TR. For sample TP, the modulus spectra at different temperatures merge into a single master curve at the unique frequency, as shown in Fig. 13a. This shows the temperature-independent behavior of the dynamic process in sample TP over the entire measured temperature range.

Scaled modulus spectra for a TP and b–d TR samples, respectively
On the other hand for sample TR, modulus spectra for an overall temperature range between 200–300 °C failed to merge into a single master curve represented in Fig. 13b. The precise analysis of modulus spectra showed that the sample can be scaled into two different ranges of temperatures, i.e., 200–240 °C and 240–300 °C displayed in Fig. 13c, d. This indicates that the sample TR shows temperature-independent characteristics only in respective ranges of 200–240 °C and 240–300 °C but not in the overall temperature range of 200–300 °C. On the basis of the above analysis, it can be concluded that the scaling behavior of modulus spectra corroborates the results obtained from the scaling nature of the conductivity spectra. The possible explanation for the conduction mechanism in the samples TP and TR could be as below.
In sample TP, Arrhenius plot (Fig. 10) shows a single slope, whereas for sample TR, there are two slopes. Similarly, for TR, two different slopes are obtained in the Arrhenius representation of the relaxation time (Fig. 12c). The single slope indicates a single conduction mechanism and double slopes indicate two different conduction mechanisms in the measured temperature range. This phenomenon for TP and TR is also confirmed in their scaling behaviors studied through conductivity and modulus formulisms. In sample TP, the conduction is thermally activated and the charge particles are participating in the conduction mechanism through the hoping. Whereas in sample TR, it appears that in the lower temperature range (200–240 °C), the charge particles are trapped in the rod-like channels, and therefore, less number of charges are participating in the conduction process. With increase in temperature (above 240 °C), the charges get enough thermal energy to liberate from the channels and all are participating in the conduction process. Thus, for TR sample, the conduction mechanism above 240 °C, the activation energy is the sum of energy required to liberate the charge particles from the rod-like structure and the energy required to hop the charge carriers.
As discussed in Sect. 3.1.2, representative high-resolution phase-contrast images acquired for TP and TR samples (with FFT as insets), as presented in Fig. 5c, d, revealed the presence of trace amount of rutile TiO2 phase in TR sample (Fig. 5e). On the other hand, in the case of TP, HRTEM analysis shows the presence of anatase TiO2 phase only and no signature of rutile TiO2 has been observed (Fig. 5c). Rutile TiO2 has a higher atomic density [55] giving rise to lower mobility to charge carriers and hence lesser conductivity [56]. The common defects in TiO2 samples are due to oxygen vacancies and Ti3+ defects. As we found from our XPS data that the area of the peak corresponding to Ti3+ is more than the peak area of Ti4+ content in sample TP. On the other hand, in sample TR, the peak area corresponding to Ti4+ is higher than the peak area of Ti3+. The higher number of Ti3+ vacancies leads to higher oxygen vacancies [35]. This may lead to higher conductivity of TP than that of TR sample. The magnitude and activation energies of both samples confirm our proposed conduction mechanism.
4 Conclusion
Two different types of nanostructures are obtained from the TiO2 nanopowders prepared via two different techniques. Sol–gel technique leads to the formation of nanoparticles and hydrothermal technique provides nanorods like structure. The XRD confirms the anatase phase formation of TiO2 with tetragonal structure and space group I41/amd. The XPS and Raman data confirm that more number of oxygen vacancies are created in the sample TP in comparative to TR. The direct bandgap was obtained as 3.19 eV and 3.25 eV for TP and TR, respectively. The scaling behavior suggests that single conduction mechanism exists in the sample TP in the entire measured temperature range, whereas, for sample TR, two different conduction mechanisms exist in the measured temperature range.
References
D.V. Wellia, Q.C. Xu, M.A. Sk, K.H. Lim, T.M. Lim, T.T.Y. Tan, Experimental and theoretical studies of Fe-doped TiO2 films prepared by peroxo sol–gel method. Appl. Catal. A Gen. 401(1–2), 98–105 (2011)
H. Sun, Y. Bai, H. Liu, W. Jin, N. Xu, G. Chen et al., Mechanism of nitrogen-concentration dependence on pH value: experimental and theoretical studies on nitrogen-doped TiO2. J. Phys. Chem. C 112(34), 13304–13309 (2008)
J. Yu, X. Zhao, Q. Zhao, Photocatalytic activity of nanometer TiO2 thin films prepared by the sol–gel method. Mater. Chem. Phys. 69(1–3), 25–29 (2001)
B. Sun, T. Shi, Z. Peng, W. Sheng, T. Jiang, G. Liao, Controlled fabrication of Sn/TiO2 nanorods for photoelectrochemical water splitting. Nanoscale Res. Lett. 8(1), 1 (2013)
L.-S. Liao, C.-H. Li, L.-L. Jiang, P.-F. Fang, Z.-K. Wang, M. Li, Enhanced electrical property of compact TiO2 layer via platinum doping for high-performance perovskite solar cells. Sol. RRL 2(11), 1800149 (2018)
H. Tang, K. Prasad, R. Sanjines, F. Levy, TiO2 anatase thin-films as gas sensors. Sens. Actuators B Chem. 26, 71–75 (1995)
D.P. MacWan, P.N. Dave, S. Chaturvedi, A review on nano-TiO2 sol–gel type syntheses and its applications. J. Mater. Sci. 46(11), 3669–3686 (2011)
V. Caratto, L. Setti, S. Campodonico, M.M. Carnasciali, R. Botter, M. Ferretti, Synthesis and characterization of nitrogen-doped TiO2 nanoparticles prepared by sol–gel method. J. Sol Gel Sci. Technol. 63(1), 16–22 (2012)
M.I. Dar, A.K. Chandiran, M. Grätzel, M.K. Nazeeruddin, S.A. Shivashankar, Controlled synthesis of TiO2 nanoparticles and nanospheres using a microwave assisted approach for their application in dye-sensitized solar cells. J. Mater. Chem. A 2(6), 1662–1667 (2014)
Y. Cheng, M. Zhang, G. Yao, L. Yang, J. Tao, Z. Gong et al., Band gap manipulation of cerium doping TiO2 nanopowders by hydrothermal method. J. Alloys Compd. 662, 179–184 (2016). https://doi.org/10.1016/j.jallcom.2015.12.034
A. Mamakhel, E.D. Bøjesen, P. Hald, B.B. Iversen, Direct formation of crystalline phase pure rutile TiO2 nanostructures by a facile hydrothermal method. Cryst. Growth Des. 13(11), 4730–4734 (2013)
A. Dey, S. De, A. De, S.K. De, Characterization and dielectric properties of polyaniline–TiO2 nanocomposites. Nanotechnology 15(9), 1277–1283 (2004)
R.J. Konwar, R. Sharma, G. Kaur, A. Mahajan, M. Kaur, P. Negi, Morpho-structural and opto-electrical properties of chemically tuned nanostructured TiO2. Ceram. Int. 44(15), 18484–18490 (2018). https://doi.org/10.1016/j.ceramint.2018.07.068
S. Takeda, S. Suzuki, H. Odaka, H. Hosono, Photocatalytic TiO2 thin film deposited onto glass by DC magnetron sputtering. Thin Solid Films 392(2), 338–344 (2001)
Z. Ding, X. Hu, P.L. Yue, G.Q. Lu, P.F. Greenfield, Synthesis of anatase TiO2 supported on porous solids by chemical vapor deposition. Catal. Today 68(1–3), 173–182 (2001)
C. Giolli, F. Borgioli, A. Credi, Fabio A. Di, A. Fossati, M.M. Miranda et al., Characterization of TiO2 coatings prepared by a modified electric arc-physical vapour deposition system. Surf. Coat. Technol. 202(1), 13–22 (2007)
G. Verma, A.K. Tripathi, M.M. Ahmad, M.K. Singh, R.K. Srivastava, A. Agarwal et al., Synthesis based structural and optical behavior of anatase TiO2 nanoparticles. Mater. Sci. Semicond. Process. 23, 136–143 (2014)
A. Yuvapragasam, N. Muthukumarasamy, S. Agilan, D. Velauthapillai, T.S. Senthil, S. Sundaram, Natural dye sensitized TiO2 nanorods assembly of broccoli shape based solar cells. J. Photochem. Photobiol. B Biol. (2015). https://doi.org/10.1016/j.jphotobiol.2015.04.017
S.H. Song, X. Wang, P. Xiao, Effect of microstructural features on the electrical properties of TiO2. Mater. Sci. Eng. B Solid State Mater. Adv. Technol. 94(1), 40–47 (2002)
D. Mardare, C. Baban, R. Gavrila, M. Modreanu, G.I. Rusu, On the structure, morphology and electrical conductivities of titanium oxide thin films. Surf. Sci. 507–510, 468–472 (2002)
R.G. Breckenridge, W.R. Hosler, Electrical properties of titanium dioxide semiconductors. Phys. Rev. 91(4), 793–802 (1953)
B. Roling, C. Martiny, S. Murugavel, Ionic conduction in glass: new information on the interrelation between the “jonscher behavior” and the “nearly constant-loss behavior” from broadband conductivity spectra. Phys. Rev. Lett. 87(8), 85901-1–85901-2 (2001)
A.K. Jonscher, The “universal” dielectric response. Nature 267, 673–679 (1977)
S. Summerfield, Universal low-frequency behaviour in the a.c. hopping conductivity of disordered systems. Philos. Mag. B Phys. Condens. Matter Stat. Mech. Electron. Opt. Magn. Prop 52(1), 9–22 (1985)
D.L. Sidebottom, Universal approach for scaling the ac conductivity in ionic glasses. Phys. Rev. Lett. 82(18), 3653–3656 (1999)
A. Ghosh, A. Pan, Scaling of the conductivity spectra in ionic glasses: dependence on the structure. Phys. Rev. Lett. 84(10), 2188–2190 (2000)
H. Tang, K. Prasad, R. Sanjinès, P.E. Schmid, F. Lévy, Electrical and optical properties of TiO2 anatase thin films electrical and optical properties of TiO2 anatase thin films. J. Appl. Phys. 75, 2042 (1994)
I.A. Alhomoudi, G. Newaz, Residual stresses and Raman shift relation in anatase TiO2 thin film. Thin Solid Films 517(15), 4372–4378 (2009). https://doi.org/10.1016/j.tsf.2009.02.141
C. Ashok, Rao K. Venkateswara, ZnO/TiO2 nanocomposite rods synthesized by microwave-assisted method for humidity sensor application. Superlattices Microstruct. 76, 46–54 (2014). https://doi.org/10.1016/j.spmi.2014.09.029
M. Razavi, M.R. Rahimipour, R. Kaboli, Synthesis of TiC nanocomposite powder from impure TiO2 and carbon black by mechanically activated sintering. J. Alloys Compd. 460(1–2), 694–698 (2008)
H.C. Choi, Y.M. Jung, S.B. Kim, Size effects in the Raman spectra of TiO2 nanoparticles. Vib. Spectrosc. 37(1), 33–38 (2005)
F. Tian, Y. Zhang, J. Zhang, C. Pan, Raman spectroscopy: a new approach to measure the percentage of anatase TiO2 exposed (001) facets. J. Phys. Chem. C 116(13), 7515–7519 (2012)
Z. Yin, W.F. Zhang, Y.L. He, Q. Chen, M.S. Zhang, Raman scattering study on anatase TiO2 nanocrystals. J. Phys. D Appl. Phys. 33(8), 912–916 (2002)
X. Pan, M.Q. Yang, X. Fu, N. Zhang, Y.J. Xu, Defective TiO2 with oxygen vacancies: synthesis, properties and photocatalytic applications. Nanoscale 5(9), 3601–3614 (2013)
B. Bharti, S. Kumar, H.N. Lee, R. Kumar, Formation of oxygen vacancies and Ti3+ state in TiO2 thin film and enhanced optical properties by air plasma treatment. Sci. Rep. 6, 1–12 (2016). https://doi.org/10.1038/srep32355
L.C. Lucas, G.N. Raikar, R. Connatser, J.L. Ong, J.C. Gregory, Spectroscopic characterization of passivated titanium in a physiologic solution. J. Mater. Sci. Mater. Med. 6(2), 113–119 (2004)
L.B. Xiong, J.L. Li, B. Yang, Y. Yu, Ti3+ in the surface of titanium dioxide: generation, properties and photocatalytic application. J Nanomater. 2012, 831524 (2012). https://doi.org/10.1155/2012/831524
V. Pawar, M. Kumar, P.A. Jha, S.K. Gupta, P.K. Jha, P. Singh, Cs/MAPbI3 composite formation and its influence on optical properties. J. Alloys Compd. 783, 935–942 (2019)
M. Kumar, V. Pawar, P.A. Jha, S.K. Gupta, A.S.K. Sinha, P.K. Jha et al., Thermo-optical correlation for room temperature synthesis: cold-sintered lead halides. J. Mater. Sci. Mater. Electron. 30(6), 6071–6081 (2019)
J.S. Lee, K.H. You, C.B. Park, Highly photoactive, low bandgap TiO2 nanoparticles wrapped by graphene. Adv. Mater. 24(8), 1084–1088 (2012)
K. Nagaveni, M.S. Hegde, N. Ravishankar, G.N. Subbanna, G. Madras, Synthesis and structure of nanocrystalline TiO2 with lower band gap showing high photocatalytic activity. Langmuir 20(7), 2900–2907 (2004)
V. Pawar, P.K. Jha, S.K. Panda, P.A. Jha, P. Singh, Band-gap engineering in ZnO thin films: a combined experimental and theoretical study. Phys. Rev. Appl. 9(5), 54001 (2018). https://doi.org/10.1103/PhysRevApplied.9.054001
P. Singh, R.K. Singh, Structural characterization, electrical and dielectric relaxations in Dy-doped zirconia. J. Alloys Compd. 549, 238–244 (2013). https://doi.org/10.1016/j.jallcom.2012.09.059
S.R. Elliott, A.C. conduction in amorphous chalcogenide and pnictide semiconductors. Adv. Phys. 36(2), 135–217 (1987)
P. Singh, O. Parkash, D. Kumar, Scaling of low-temperature conductivity spectra of BaSn1– xNbxO3 (x ≤ 0 100): temperature and compositional-independent conductivity. Phys. Rev. B. 84(17), 174306 (2011). https://doi.org/10.1103/PhysRevB.84.174306
N.K. Singh, P. Singh, M.K. Singh, D. Kumar, O. Parkash, Auto-combustion synthesis and properties of Ce0.85Gd0.15O1.925 for intermediate temperature solid oxide fuel cells electrolyte. Solid State Ion. 192(1), 431–434 (2011). https://doi.org/10.1016/j.ssi.2010.04.015
B. Roling, A. Happe, K. Funke, M.D. Ingram, Carrier concentrations and relaxation spectroscopy: new information from scaling properties of conductivity spectra in ionically conducting glasses. Phys. Rev. Lett. 78(11), 2160–2163 (1997)
A.K. Yadav, P.A. Jha, S. Murugavel, P. Singh, Synthesis, characterization and AC conductivity of alkali metal substituted telluride glasses. Solid State Ion. 296, 54–62 (2016). https://doi.org/10.1016/j.ssi.2016.08.013
M. Khutia, G.M. Joshi, S. Bhattacharya, Study of electrical relaxation mechanism of TiO2 doped Bi-polymer systems. Adv. Mater. Lett. 7(3), 201–208 (2016)
S. Murugavel, B. Roling, Ion transport mechanism in borate glasses: influence of network structure on non-Arrhenius conductivity. Phys. Rev. B Condens. Matter Mater. Phys. 76(18), 2–5 (2007)
D.N. Singh, T.P. Sinha, D.K. Mahato, Electric modulus, scaling and ac conductivity of La2CuMnO6 double perovskite. J. Alloys Compd. 729, 1226–1233 (2017). https://doi.org/10.1016/j.jallcom.2017.09.241
P.A. Jha, A.K. Yadav, P.K. Jha, P. Singh, AC conductivity and ion dynamics of alkaline earth metal substituted telluride glasses. J. Non Cryst. Solids 452, 203–209 (2016). https://doi.org/10.1016/j.jnoncrysol.2016.08.043
O.N. Verma, N.K. Singh, P. Singh, Study of ion dynamics in lanthanum aluminate probed by conductivity spectroscopy. RSC Adv. 5(28), 21614–21619 (2015). https://doi.org/10.1039/C5RA01146A
P. Singh, B.P. Singh, Dispersion in AC conductivity of fragile glass melts near glass transition temperature. Solid State Ion. 227, 39–45 (2012). https://doi.org/10.1016/j.ssi.2012.08.021
D.A.H. Hanaor, C.C. Sorrell, Review of the anatase to rutile phase transformation. J. Mater. Sci. 46, 855–874 (2011). https://doi.org/10.1007/s10853-010-5113-0
M.K. Nowtony, T. Bak, J. Nowtony, Electrical properties and defect chemositry of TiO2 single crystal. I. Electrical conductivity. J. Phys. Chem. (2006). https://doi.org/10.1021/jp0606210
Acknowledgements
Authors are thankful to Dr. S. K. Gupta (Department of Physics, Banasthali Vidyapeeth, Banasthali, Rajasthan) for providing Raman measurement facility.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pawar, V., Kumar, M., Dubey, P.K. et al. Influence of synthesis route on structural, optical, and electrical properties of TiO2. Appl. Phys. A 125, 657 (2019). https://doi.org/10.1007/s00339-019-2948-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-019-2948-3