
Abstract
Li+–Er3+/Yb3+ co-doped Y2O3 upconversion films were prepared via a sol–gel method and spin coating. Li+ doping resulted in better crystallinity and larger crystal grains. All the films had purple, green and red upconversion emission bands. Li+ doping decreased the lattice symmetry of the matrix and enhanced energy transfer, and green and red emissions improved by more than 100 times. Meanwhile, Li+ doping also increased the lifetime of both the green and red emissions significantly via reducing the probability of non-radiation transition. Moreover, the 4S3/2 energy level tends to be populated with increasing Li+, the matrix phonon energy change and the fluorescence quenching causing it. When the mole ratio of rare earth ions to Li+ was over 1:2, the purple emission shifted from 436 to 409 nm. Here, Li+ played a switching role in the conversion of these two channels. Li+ doping up to a critical concentration value changed the matrix phonon energy. The match of the phonon energy with the energy gap between 2G7/2–2H9/2 and that of 2G7/2–4F3/2 was the reason.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent years, upconversion (UC) luminescent materials have been widely used in many fields such as optical thermometry [1], solid-state lighting [2], fingerprint detection [3], imaging [4], iatrology [5] and radiology [6]; so, they have received attention. Especially, in the development of UC materials, rare earths (RE)-doped luminescent materials are remarkable [7,8,9,10]. Due to the special 5s25p6 shell, which is a shell blocking 4fN, RE ions have many good fluorescence characteristics, such as pure luminance, high conversion efficiency, a wide range of emission wavelength and stable material properties [11]. Therefore, RE-doped UC materials are used in the fields of fluorescent equipment [8, 12], lasers [13, 14], waveguide laser [15], etc. Particularly, thin films have become the focus of research due to their benefits of small volume [16], large surface area [17] and stable chemical properties [18].
UC materials are composed of an activator ion (luminescence centre), sensitizer ion, matrix and other doped ions. The Er3+ ion [19,20,21] as a common activator ion is widely used in many fields due to its two important advantages. First, the rich energy levels of the Er3+ ion produce rich emissions, which include the purple emission at 400–440 nm, the green emission at 520–580 nm, the red emission at 640–700 nm and the near-infrared emission at 1520–1580 nm. This feature allows Er3+ ion to fit multiple optical devices. Second, the Er3+ ion can absorb excitation light at 980 nm without sensitizer ions and jump to the 4I11/2 energy level directly. Therefore, it can luminesce directly on its own to incorporate multiple ions [22]. Yb3+, as a sensitized ion, is often paired with Er3+ ion [23] and has the following three advantages for sensitization. First, the energy levels of Yb3+ are simple and independent, which means that the ground state absorption (GSA) process is simple, and the transition energy level is fixed [24, 25]. As a result, there is less energy loss when the energy transmits from Yb3+ to Er3+. Second, the position of the Yb3+ ion’s 2F5/2 level is close to that of the Er3+ ion’s 4I11/2 level, which means that the Yb3+ ion’s energy can easily transmit to Er3+. Third, Yb3+ ions can be activated with a 980-nm or 488-nm pump laser; also, it has a large absorption cross section, which is beneficial to improve UC efficiency. For the selection of the matrix, it is best to choose low phonon energy materials, such as fluoride [8, 26] and other halides [27]. However, these compounds are not suitable for making films due to poor chemical stability and toxicity. Therefore, oxide, which has good chemical stability, moderate phonon energy and is nontoxic, is a good option [28,29,30]. Especially, Y2O3 has a crystal structure similar to the RE oxides, leading to the RE3+ ions easily doping into it, which has aroused general interest in the last decade [31, 32].
Since the UC process was discovered in the 1960s [33,34,35], improving the efficiency of the UC has always been the research direction and focus. Li+ is highly suitable to be incorporated into the host and modify the crystal field around the RE ions in such a way that it improves the luminescent efficiency. The mechanism is that doping with Li+ ions can tailor the local crystal field around RE3+ ions by substituting host cations or occupying the interstitial lattice sites. The tailored local crystal field can interact with the RE ions, slightly modify their wave functions, alter the radiation properties of the intermediate state and finally increase the visible UC radiation. Ramakrishna et al. [36] studied the structural and photoluminescence behaviour of thermally stable Eu3+-activated CaWO4 nanophosphors via Li+ incorporation; the results showed that Li+ enhanced the crystallinity and radiative transition rate, thus resulting in higher emission. Yan et al. [37] synthesized Li+-doped GdVO4:Eu3+ nanophosphors by an efficient hydrothermal method. The results demonstrated that Li+ doping changed the lattice parameters in two different ways, and Li+ ion also increased the concentration quenching threshold of Eu3+. Wang Xiuwen et al. [38] synthesized Li–NaYF4:Yb/Er nanocrystals and performed a comprehensive investigation of the effect of Li+ doping on the morphology, crystal structure, and UC luminescence. They found that Li+ doping led to the crystal lattice changing from hexagonal to a mixture of cubic and tetragonal, and finally to pure tetragonal. In addition, 0.5 mol% Li+ doping was the most efficient, which enhanced green emission 34 times and red emission 101 times. Wang Yangbo et al. [39] prepared Li+-doped KSc2F7:Yb/Er nanocrystals. Li+ doping induced local crystal field variation around the doped lanthanide ions and resulted in a 21-fold red luminescence enhancement and 22-fold green luminescence enhancement. Meza-Rocha et al. [40] prepared Y2O3:Er3+ films and found that the green and red emissions were improved up to 365 and 171 times the original when Li+ doped. Avram Daniel et al. [41] extensively investigated the evolution of Li addition and calcination temperatures of the down-conversion (optical and X-ray induced) and UC emission of Ln–Y2O3 measured across the visible to near-infrared range. Li+ enhanced the emissions markedly, and they found that the reason for the enhancement was that Li+ not only reduced the symmetry of the Y2O3 lattice, but also improved crystallization. In addition, these studies have elaborated the mechanisms of the enhancement of luminescence by Li+. However, there are still two issues requiring further discussion: first, whether all the emissions increase in the same proportion when the concentration of Li+ increases; second, how the emissions in the short wave region (under 500 nm) change in the Yb3+/Er3+ system with Li+ doping.
To investigate the two problems above, the Li+ and Yb3+/Er3+ co-doped Y2O3 UC films were prepared via a sol–gel and spin-coating technique. The UC films were characterized with multi-functional field-emission scanning electron microscopy (SEM), energy-dispersive spectrometry (EDS), X-ray diffractometry (XRD), Fourier transform infrared spectroscopy (FTIR), and photoluminescence spectra (PL). The effects of Li+ doping on the structure and property of the UC films were investigated.
2 Experimental details
2.1 Films preparation and molar fractions of the ions
The Y2O3 (with Yb3+, Er3+ and Li+) precursor was prepared using the sol–gel method. All chemicals were used directly without further purification. In a typical synthesis, the stoichiometric composition of Y2O3 (3N), Yb2O3 (3N), Er2O3 (3N), and LiNO3 (3N) materials were mixed and dissolved in a certain concentration of nitric acid. Finally, citric acid was added and then stirred at 80 °C. The precursor sol was aged at room temperature for 24 h before spin coating. The contents of ions are shown in Table 1. The Y3+ content was set as the standard, and the other ions were added according to that benchmark. The samples were named after the mole ratio of RE3+:Li+.
Quartz glass was chosen as the substrate due to its excellent optical transmittance and high thermal stability. The size of each glass was 50 mm × 50 mm. All substrates were cleaned with ethanol and deionized water, then corroded to increase the surface roughness with concentrated sulfuric acid and strong ammonia water, and finally cleaned ultrasonically for 20 min. The substrates were dried in an oven after cleaning with ethanol and deionized water again. Then, the sol was deposited onto the glass substrates with spin coating and then dried to form films. Finally, for the thermal process the samples were dried in air at 200 °C for 30 min, followed by an annealing process at 1000 °C for 2 h.
2.2 Characterization and optical measurements
The visible and near-infrared transmittance of each film was measured with a UV-3600 spectrophotometer. The film’s morphology was observed with SEM and its chemical composition determined with EDS; both of these were done on the same equipment, a Hitachi S4800 FE-SEM. The crystal structure of each film was characterized with XRD, a BRUKER D 8 DISCOVER system. The FTIR spectra of the films were measured with a Nicolet iS50 instrument in the 400–3000 cm− 1 wave number range. The PL spectrum of each film was obtained with a Hitachi F-4500 instrument pumped with a 980 nm laser. The UC luminescence decay of each film was measured with Edinburgh instruments FLS 1000 spectrometer.
3 Results and discussion
3.1 Transmittances and surface morphology of thin films
Figure 1 shows the transmittance of the Li+–Yb3+/Er3+ co-doped Y2O3 UC films. It is clear that all the samples have excellent transmittance, higher than 97% in the 350–800 nm range. Obviously, the curves in Fig. 1 can be indexed to two groups. Group I includes the samples 1:0, 1:1, 1:1.5, 1:2 and 1:3. The other, 1:2.5, belongs to group II. All the curves in group I first decrease strongly from 350 nm to 400 nm, then decrease slowly with the further increase of wavelength. The order of the transmittances from low to high is 1:1.5 < 1:1 < 1:2 < 1:3 < 1:0. In addition, the overall changes are small (under 3%). The 1:2.5 sample represents group II; its curve keeps increasing with the increase of the wavelength. In general, Li+ doping reduces the film transmittance, but the reduction is very limited.

The transmittance of Li+–Er3+/Yb3+ co-doped Y2O3 films (sample 1:0 to 1:3)
The UC films’ surface topographies are shown in Fig. 2a, b. Figure 2a shows the film surface of sample 1:0. The film surface is composed of nanoparticles with unequal distributions. Grains are generally round, smooth, full and less aggregated. Thus, the film crystallized well, and the nanoparticle sizes are approximately 30–60 nm in the images.

The SEM images of Er3+/Yb3+ co-doped Y2O3 upconversion films. a Sample 1:0 (without Li+ doping), b sample 1:2.5 (with Li+ doping)
Because the five samples doped with Li+ are nearly the same, 1:2.5 is chosen to be representative of characterizing the surface of the Li+-doped films, for which the SEM image is shown in Fig. 2b. As observed from the image, the film surface is also composed of nanoparticles, which crystallized better and larger than those of sample 1:0; some of them even aggregated to form some embossments. According to the scale, their sizes are estimated to be 50–110 nm. Therefore, it could be said that Li+ ion doping brought about grain agglomeration and embossments on the surface. This could be used to explain the transmittance curves in Fig. 1. These embossments increased the light scattering and decreased the film transmittance, though the better crystallization might improve the film’s transmittance to some extent. In general, Li+ doping decreased the film’s transmittance a little.
3.2 Thin film compositions and crystal structure
When the film surface was observed with SEM, its chemical compositions were also analysed with EDS. All the samples’ test outcomes were nearly the same. Therefore, the chemical compositions of sample 1:2.5 are shown in Fig. 3 as a typical example. The Y3+ characteristic peak at 2.0 keV is the highest, which means that the main phase should be Y2O3. The peaks of Yb3+ and Er3+ appear between 7.0 and 9.0 keV. However, the Li+ characteristic peak has not been highlighted because the energy of Li+ in the X-ray spectrum is lower than that of the equipment’s window material (Beryllium), which hid the Li+ characteristic peak in the background noise. This can also explain why all the test results are almost identical.

EDS spectra of Li+–Er3+/Yb3+ co-doped Y2O3 film (sample 1:2.5)
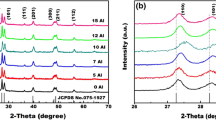
Figure 4a shows the XRD patterns of the Y2O3 thin films doped with Li+, Er3+ and Yb3+. All the patterns are similar, and all the diffraction peaks are clear and sharp, which means all the samples crystallized well. In addition, all the main diffraction peaks, (211), (222), (400), (440) and (622), could be indexed to the characteristic peaks of a Y2O3 crystal. This suggests that no new crystal phase formed after the RE3+ and Li+ doping, and the vast majority of the doping ions entered into the Y2O3 crystal lattice. Moreover, the positions of the main diffraction peaks (222) can be observed to shift to a lower angle. The main diffraction peaks (222) of the XRD patterns are magnified in Fig. 4b. The inset of Fig. 4b shows the shifts of the main diffraction peaks more clearly.

XRD patterns of Li+–Er3+/Yb3+ co-doped Y2O3 flims and the standard card of Y2O3 (PDF, No. 41-1105). a Effect of Li+ ion on the XRD patterns of the films (Sample 1:0 to 1:3); b the shift of the main diffraction peaks (222) of the XRD patterns (sample 1:0 to 1:3)
Herein, the average lattice constants were calculated with Bragg’s Law, 2dsinθ = nλ, where d is the interplanar crystal spacing, θ is the angle between the incident X-ray and crystal face, n is the series of diffraction, and λ is the X-ray wavelength 0.154056 nm. The average crystallite size of each film was calculated with Debye–Scherrer formula, D = kλ/(βcosθ) [42], where D is the average grain size, λ the X-ray wavelength 0.154056 nm, θ the Bragg angle of diffraction peak, k the Scherrer constant conventionally set to be 0.89, and β is the corrected FWHM (full width at half maximum) of the main characteristic diffraction peak. The positions of the main diffraction peaks (222), their FWHMs, the average lattice constants, and the average crystallite sizes of the films are listed in Table 2. The FWHM decreases and the average crystallite size increases with Li+ concentration increase. This supports the result from Fig. 2 mentioned above and is consistent with the results of Avram Daniel et al. [41].
As exhibited in the inset of Fig. 4b, the vertical solid line is the peak (222) of the standard card, and the dotted line on the right, (222)′ is set as the standard to discuss the effect of the Li+ ion. At the main peak position (222) and the average lattice constant, RE ion doping increased the main peak diffraction angle while decreasing the average lattice constant at first. Then, Li+ doping resulted in the left shift of the main peak position (222) and the increase of the average lattice constant in general. However, neither the shift nor the increase was proportional to the Li+ concentration, or even a unilateral increase.
Because the radius of Li+ (0.76 Å) is smaller than that of Y3+ (0.89 Å), the doped Li+ might replace Y3+ in the lattice, which would shrink the interplanar spacing, d, leading to a θ increase according to Bragg’s Law, i.e. the peak shifting right. If Li+ enters the lattice interstitially, the lattice will swell, leading to θ decrease, i.e. the peak shifting left. From Table 2, it could be inferred that the doped Li+ initially entered into the Y2O3 crystal lattice interstitial sizes, resulting in the lattice constant increase for samples 1:0 and 1:1. Next, additional Li+ ions might have replaced Y3+ ions in the lattice, resulting in the lattice constant decrease for samples 1:1, 1:1.5, and 1:2. With further concentration increase, more Li+ ions might have entered the Y2O3 crystal lattice interstitial sizes, and the lattice constant increased again for samples 1:2, 1:2.5, and 1:3. Finally, the lattice constant of sample 1:2.5 is closest to that of the pure Y2O3 powder.
FTIR spectroscopy carries rich information about molecular vibrations. To investigate the parasitic adsorption, the FTIR spectra of the samples are collected and shown in Fig. 5. Each curve has strong sharp absorption peaks at 473 cm− 1 and 566 cm− 1. The peak at 473 cm− 1 arises from the asymmetric stretching vibrations of the Y–O bond and the RE–O bond; the peak at 566 cm− 1 is ascribed to the symmetric stretching vibrations of the Y–O–Y bond and the RE–O–RE bond. A weak absorption peak occurs at 880 cm− 1 and becomes stronger with the increase of the concentration of Li+ ions. This peak comes from the stretching vibration of the O–O bond, which is a defect of the crystal. Sample 1:0 does not have this peak. This peak becomes larger with the increase in concentration of Li+. The main reason should be the addition of Li+. The valence of Y3+ is different from that of Li+. When Li+ ions enter the lattice, whether by substitution or in an interstitial manner, the original charge balance is broken. The O–O bond appears to balance the charge of the crystal lattice. More Li+ ions bring more O–O bonds, so this peak becomes deeper as the Li+ concentration increases. In other words, the number of the crystal surface defects [43] increases. However, even in sample 1:3, the 880 cm− 1 peak is still extremely weak, which means the crystal surface defects are not too significant and do not play a major role in UC luminescence.

FTIR spectra of Li+–Er3+/Yb3+ co-doped Y2O3 films (sample 1:0 to 1:3)
3.3 UC luminescence
Figure 6 illustrates the UC spectra of the films in the range of 400–700 nm excited with a 980-nm diode laser at a constant power of 1 W.

PL spectra of Li+–Er3+/Yb3+ co-doped Y2O3 films (sample 1:0 to 1:3, λex = 980 nm); the inset is the zoom of the peaks in the purple region (400–450 nm)
There are three emission bands in every spectrum, including a purple one located in the range of 400–450 nm ascribed to 2H9/2/4F3/2 → 4I15/2 of Er3+, a green one located at 520–580 nm ascribed to 2H11/2/4S3/2 → 4I15/2 of Er3+ and a red one located at 630–680 nm ascribed to 4F9/2 → 4I15/2 of Er3+. The emission intensities of all the samples are much stronger in the green and red regions than at purple.
The inset of Fig. 6 shows the emissions in the purple region. As shown in the figure, when the concentration of Li+ is low, the emission appears at 436 nm or disappears. However, once the Li+:RE3+ ratio is over 2:1 (samples 1:2.5 and 1:3), the emission peak shifts left to 409 nm. The 409 nm emission and 436 nm emission are mutually exclusive. Here, the Li+ ion plays the role of a photo-switch to some extent. For the green and red emission, the emission intensities increase with the increase of Li+ concentration. In more detail, this kind of increase is very limited at first when Li+ concentration is low. Once the Li+:RE3+ ratio is over 2:1, both the green and red emissions will improve greatly to more than 100 times the original intensity. The enhancements are ascribed to both the lattice asymmetry increase and the improved crystallinity mentioned above.
The peak intensities of the UC spectra in these three areas are shown in Fig. 7a–c, respectively, for us to further discuss the photo-switch effect in the purple region and the energy population change in the green and red regions. Figure 7a shows that when the sample is not doped with Li+ or doped with a low concentration, the emission is very weak and could even be neglected, and the peak appears at 436 nm, which can be ascribed to 4F3/2 → 4I15/2 of Er3+. However, once the concentration of Li+ is twice as much as that of RE3+, the emission is enhanced and the peak moves to 409 nm, which can be ascribed to 2H9/2 → 4I15/2 of Er3+. Therefore, it could be said that the two emissions are mutually exclusive. Here, Li+ ion concentration played a switching role in the conversion of these two emission channels, 4F3/2 → 4I15/2 and 2H9/2 → 4I15/2, which could be called a photo-switch effect.

a The main peaks in the purple region of the UC spectra, b the main peaks in the green region of the UC spectra, and c the main peaks in the red region of the UC spectra
Figure 7b, c reveals the change in energy populations. As shown in Fig. 6, there are four peaks in every curve in the green region. The left two are ascribed to 2H11/2 and its Stark level of Er3+, and the emission at 535 nm is chosen to be the representative. The right two are ascribed to 4S3/2 and its Stark level of Er3+, and the emission of 535 nm is chosen to be the representative. The intensity of these two emission peaks varies with the concentration of Li+, as shown in Fig. 7b. When the Li+ concentration is within 1:0 to 1:2, these two curves show similar tendencies; the emission intensity first increases, then decreases, and finally increases again. As the Li+ concentration increases from 1:2 to 1:2.5, the intensity increases approximately 100 times. However, then the tendency becomes different. The 556 nm emission intensity continues to increase, while the 535 nm emission intensity decreases when the Li+ concentration increases from 1:2.5 to 1:3. As shown in Fig. 7c, the intensity of the red emissions of 660 nm and 673 nm ascribed to 4F9/2 and its Stark sublevel 4I15/2 of Er3+ present the same change tendency. In general, the red emission intensity first increases very slowly with the Li+ concentration increase from 1:0 to 1:2, then increases dramatically from 1:2 to 1:2.5, and next decreases rapidly.
In brief, Li+ doping can enhance the green and red emissions significantly. Li+ doping concentration plays a photo-switch role to control the mutually exclusive 409 nm and 436 nm emissions in the purple band. Moreover, an interesting turning point in Fig. 7b should be paid more attention. The intensities of 535 nm green emission, and 660 nm and 673 nm red emission all decrease while the intensity of 556 nm green emission still increases as the Li+ concentration increases from 1:2.5 to 1:3. This means the more excited Er3+ populate the 4F9/2 level, which reveals two points: first, the excited Er3+ tends to populate the 4S3/2 level; second, fluorescence quenching occurs.
The UC decays of the films were measured for us to further study the luminescence mechanism. According to the fitting formula [44]: \(R(t)=A+{B_1}\exp ( - t/{\tau _1})+{\text{ }}{B_2}\exp ( - t/{\tau _2})+ \cdots ,\) where R(t) is the intensity of the luminescence; τ1 and τ2 are fluorescence lifetime; t1 and t2 are decay times; A, B1 and B2 are constants; \(\tau =\frac{{\text{1}}}{{1/{\tau _1}+1/{\tau _2}}}\); and the fitting curves are drawn in Fig. 8. The results show that all of these intensity decays can be satisfactorily fitted with a double exponential decay model. The recovered luminescence lifetimes, corresponding fractions, and the average lifetimes are listed in Table 3.

UC luminescence decay curves of Li+–Er3+/Yb3+ co-doped Y2O3 films; a decay profiles of the UC luminescence at 556 nm, the inset is the zoom at the range of 650–850 µs; b decay profiles of the UC luminescence at 660 nm, the inset is the zoom at the range of 600–750 µs
As shown in Fig. 8a, Li+ ion doping increases the upconversion green emission lifetime of the film significantly. Seen from the inset figure, further Li+ doping does not increase the fluorescence lifetime to a great degree. As the results mentioned in Fig. 6 show, the green emissions are ascribed to 4S3/2 → 4I15/2. The fluorescence life is inversely proportional to the sum of the probability of radiation transition and the probability of non-radiation transition of the energy level [45]; therefore, the fluorescence life τ can be: expressed as τ = 1/(A + W), where A is the probability of radiation transition; W is the probability of non-radiation transition. Seen from Table 3, the average decay time for green emission increases 1.59–1.89 times after Li+ ion doping. Seen from Fig. 6, Li+ ion is the only factor to enhance the emission, which means that Li+ ion doping reduces the probability of non-radiation transition distinctly. Figure 8b shows a similar pattern to Fig. 8a; the average decay time for red emission increases 1.31–1.55 times after Li+ ion doping, which indicates that Li+ ions reduce the probability of non-radiation transition in an overall way of the energy transition.
For a better understanding of the UC process, the pump power dependence of the UC emission was measured. For small UC version rates, the luminestcence intensity, IUC, follows the relation \({I_{{\text{UC}}}} \propto {P^n}_{{{\text{pump}}}}\) [46], where Ppump is the incident pump power and n is the number of photons required to populate the emitting state. The plot of IUC versus Ppump with a double logarithmic scale yields a straight line with a slope n. Figure 9a shows a logarithmic plot of the integrated emission intensity of the UC luminescence as a function of pump power for the Li+–Yb3+/Er3+ co-doped Y2O3 UC film (sample 1:2.5) in every emission band. The 409 nm emission was chosen to be representative of the purple band and the slope was 2.918, which means that the purple emission was a three-photon process. Likewise, the 556 nm and 660 nm emissions were also chosen to be representatives of green and red emissions, respectively. Their slopes n are 1.971 and 1.784 for the 556 nm and 660 nm emissions, respectively. These results indicate that two-photon processes contribute to the UC of the green and red emissions. The Yb3+ ions and a small number of Er3+ ions absorb 980 nm photons and transfer their energy to both the major Er3+ ions and the Y2O3 matrix.

a PL spectra of Li+–Er3+/Yb3+ co-doped Y2O3 film in different pump laser power (sample 1:2.5), b log–log plots of Li+–Er3+/Yb3+ co-doped Y2O3 film (sample 1:2.5, λem = 409, 556 and 660 nm)
3.4 The mechanisms of energy transfer and UC emissions
The energy-level diagram of the Er3+–Yb3+ ion pair is depicted in Fig. 10a. On one hand, Yb3+ ions act as highly effective sensitizers for Er3+, producing the anti-Stokes emissions corresponding to the 4f → 4f transitions. In the first step, the Yb3+ ions are excited from the 2F7/2 to the 2F5/2 level via absorbing photons at 980 nm. Then, the Er3+ ions transfer from the 4I15/2 to the 4I11/2 level via energy transition from the Yb3+ ions at the 2F5/2 level (ET1). In the next step, Er3+ ions at the 4F7/2 level will be populated via energy transfer (ET2) of 2F5/2(Yb) + 4I11/2(Er) → 2F7/2(Yb) + 4F7/2(Er). Then, a portion of Er3+ ions at the 4F7/2 level undergoes nonradiative decay to the nearest lower level, 4H11/2, and its Stark level. Then, a great portion of the excited Er3+ ions radiatively decay from the 4H11/2 level to the ground level, generating two strong green emissions at approximately 535 nm (the left two peaks in the green region in Fig. 6). Meanwhile, another portion of Er3+ ions at the 4F7/2 level undergo nonradiative decay to 4S3/2, which is under 4H11/2 and its Stark level, then drop to the ground, generating two more strong green emissions at approximately 556 nm (the right two peaks in the green region in Fig. 6).

Energy-level diagram of the Li+–Yb3+/Er3+-doped Y2O3 film as well as the UC mechanisms; Excitation wavelength: 980 nm. a The mechanisms of the full process, b the mechanism of the photo-switch effect
At the same time, the third portion of Er3+ ions at the 4F7/2 level undergo nonradiative decay to a slightly lower level, 4F9/2, introducing heat to the host lattice. Then, a red emission at approximately 660 nm originates from the 4F9/2→4I15/2 radiative transition of Er3+ ions. The second path for the red emission at approximately 660 nm is as follows. A portion of Er3+ ions are excited to the 4I11/2 level from ET1 decay to the 4I13/2 level via multiphonon relaxation process. Then, the energy transfer (ET3) of 2F5/2(Yb) + 4I13/2(Er) → 2F7/2(Yb) + 4F9/2 (Er) will occur, which makes Er3+ ions populate the 4F9/2 level. On the other hand, a small number of Er3+ ions, acting as the sensitized ions similar to Yb3+, are excited from the 4I15/2 to the 4I11/2 level via absorbing photons at 980 nm. Some of them transform into luminescence centres, decay to level 4I13/2 and join the ET3 process. The other Er3+ ions at level 4I11/2 rise to the Stark sublevel of 4F9/2 via cross relaxation (CR), with Er3+ ions at level 4F7/2 dropping to level 4F9/2. Then, the red emissions at 660 nm and 673 originate from the 4F9/2 and its Stark sublevel to 4I15/2 in a radiative transition of the Er3+ ions. This is the third path for the red emission.
Doping Li+ into the Y2O3:Er3+, Yb3+ thin film can boost UC luminescence intensity, as Li+ ions embedded in the Y2O3 lattice increase the asymmetry of the crystal field via replacement or in an interstitial manner. Meanwhile, the doped Li+ ions also increase the grain size. These two points are the main mechanism of Li+ enhancing the UC luminescence discussed in Sects. 3.2 and 3.3, which are also confirmed by other works [41, 47,48,49,50,51,52].
In this work, the UC enhancement degree from Li+ doping for each emission band is different. In other words, Li+ doping also affects the energy state population. When the ratio of RE3+:Li+ is less than or equal to 1:2, the increase of each peak intensity is almost in an equal proportion, which means that the energy population does not change. However, when the ratio of RE3+:Li+ reaches 1:2.5, the red emission enhancement is larger than that of the green one, which means that the excited Er3+ ions tend to populate at the 4F9/2 level. The reason is that an appropriate amount of Li+ ions enhance the energy transfer, leading to additional excited Er3+ ions populating the 4F7/2 level; then, the CR process is enhanced, and the 4F9/2 level stores more Er3+ ions than before. Finally, the red emission receives a greater boost. Compared to sample 1:2.5, the 556 nm emission is still increased, but the 535 nm and the red emissions start to fall back, which means that the number of excited Er3+ ions at 4S3/2 still increase, but the others start decreasing. In other words, the 4S3/2 level tends to be populated. Two factors contribute to this situation. First, Li+ doping not only breaks the Y2O3 lattice symmetry, but also changes the matrix phonon energy, and all the bridges between the energy levels are built by multiple phonons. Compared with the gap between 4F7/2 and 2H11/2, the phonon energy of sample 1:3 is more compatible with the gap between 4F7/2 and 4S3/2 obviously. Therefore, the 556 nm emission still increases, but the 535 nm emission decreases. Second, compared with sample 1:2.5, the total emission intensity of sample 1:3 decreases, and fluorescence quenching occurs and leads to the CR process weakening. Therefore, the number of Er3+ ions on the 4F9/2 level decreases, and then the 660 nm and 673 nm emissions recede.
To concisely expound the mechanism of the photo-switch effect, the energy transfer process of the purple band is depicted in Fig. 10b. The excited Er3+ ions at the 2H11/2 level jump to 2G7/2 via the energy transfer (ET4) of 2F5/2(Yb) + 4F7/2(Er) → 2F7/2(Yb) + 2G7/2(Er), then decay to the lower levels 2H9/2 and 4F3/2 and finally drop to the ground level; thus, the emissions at 409 nm and 436 nm occur. As shown in Fig. 8, the purple emission is a three-photon process occurring with much more difficulty than a two-photon process. Compared with the green and red emissions, the emissions in the purple region are significantly less intense. The energy is very easy to lose, so the bridge is more suitable. The photo-switch effect between the 409 and 436 nm emissions is also caused by the phonon energy. The energy gap between 2G7/2 and 2H9/2 might be closer to the integral multiple of the phonon energy of samples 1:2.5 and 1:3. For samples 1:0, 1:1.5 and 1:2, their multiple phonon energy might not fit the gap between 2G7/2 and 2H9/2 well, but might match the gap between 2G7/2 and 4F3/2 better. In addition, sample 1:1 does not match any energy gap. Predictably, the excited ions at the higher energy level will be more isolated.
4 Conclusions
A series of Li+–Er3+/Yb3+ co-doped Y2O3 UC films with various concentrations of Li+ ions were prepared via a sol–gel method and spin coating. The Li+ ions’ photo-switch effect and mechanism affecting the energy-level populations were investigated.
All the films were composed of nanoparticles and showed high transmittance more than 97%; the doped Li+ ions entered the Y2O3 lattice via replacement or in an interstitial manner. Li+ doping resulted in larger crystal grains, reducing the film’s transmittance a little.
Excited with a 980-nm diode laser, all the films had purple, green and red upconversion emission bands. Li+ doping decreased the lattice symmetry of the matrix and improved energy transfer, so both green and red emissions improved more than 100 times. Also, Li+ doping also increased the lifetime of both the green and red emissions significantly via reducing the probability of non-radiation transition. Moreover, the 4S3/2 energy level tended to be populated to increase the 556 nm emission with the Li+ concentration increase, which could change the matrix phonon energy and depress the fluorescence quenching.
When the ratio of the rare earth ion to Li+ was over 1:2, the purple emission shifted from 436 nm to 409 nm. This is because the energy level transition changed from 4F3/2–4I15/2 to 2H9/2–4I15/2. Here, Li+ played a switching role in the conversion of these two emission channels, which could be called a photo-switch effect. The mechanism is that Li+ doping up to a critical concentration value changed the matrix phonon energy and thus improved the match of the phonon energy with the energy gap between 2G7/2–2H9/2, while the match with the energy gap between 2G7/2–4F3/2 became poorer. Both of these two energy matches are necessary for the purple emission, a three-photon upconversion.
References
P. Du, L. Luo, J.S. Yu, Upconversion emission and cathodoluminescence of Er3+ -doped NaYbF4 nanoparticles for low-temperature thermometry and field emission displays. Appl. Phys. A 123, 157 (2017)
P. Du, J.S. Yu, Near-infrared light-triggered visible upconversion emissions in Er3+/Yb3+-codoped Y2Mo4O15 microparticles for simultaneous noncontact optical thermometry and solid-state lighting. Ind. Eng. Chem. Res. 57, 13077–13086 (2018)
P. Du, P. Zhang, S.H. Kang, J.S. Yu, Hydrothermal synthesis and application of Ho3+-activated NaYbF4 bifunctional upconverting nanoparticles for in vitro cell imaging and latent fingerprint detection. Sens. Actuator B Chem. 252, 584–591 (2017)
Z. Zhang, Y. Zhang, C. Wang, Z. Feng, W. Zhang, H. Xia, White light emission characteristics of Tb3+/Sm3+ co-doped glass ceramics containing YPO4 nanocrystals. J. Mater. Sci. Technol. 33, 432–437 (2016)
Y. Jiang, H. Xia, J. Zhang, S. Yang, H. Jiang, B. Chen, Growth and optical spectra of Tb3+/Eu3+ co-doped cubic NaYF4 single crystal for white light emitting diode. J. Mater. Sci. Technol. 31, 1232–1236 (2015)
E.J. Majdak-Paredes, M.V. Schaverien, P. Szychta, C. Raine, J.M. Dixon, Intra-operative digital specimen radiology reduces RE–operation rates in therapeutic mammaplasty for breast cancer. Breast 24, 556–559 (2015)
H.H. Cheng, W. Lin, Z.Q. Luo, Z.M. Yang, Passively mode-locked Tm3+-doped fiber laser with gigahertz fundamental repetition rate. IEEE J. Sel. Top. Quantum Electron. 24, 6 (2018)
Z.C. Yang, X.D. Wang, Y.C. Wu, X.F. Li, M.S. Chen, Two emission turn-on optical chemosensors for cysteine detection using up-conversion nanocrystals as excitation host: synthesis, characterization and performance. Sens. Actuator B Chem. 255, 1587–1594 (2018)
W.A. Pisarski, J. Janek, J. Pisarska, R. Lisiecki, W. Ryba-Romanowski, Influence of excitation wavelengths on up-converted luminescence sensing behavior of Er3+ ions in lead-free germanate glass. J. Lumin. 193, 34–38 (2018)
J.L. Qin, C.F. Hu, B.F. Lei, J.F. Li, Y.L. Liu, S.P. Ye, M.Z. Pan, Temperature-dependent luminescence characteristic of SrSi2O2N2:Eu2+ phosphor and its thermal quenching behavior. J. Mater. Sci.Technol. 30, 290–294 (2014)
L.W. Jiang, Z.H. Wang, Y.P. Chen, P. Chen, L.H. Luo, H.B. Chen, Bright up-conversion emission of Er3+-doped lead-free ferroelectric Na0.5Bi0.5TiO3 single crystal. Mater. Lett. 210, 158–160 (2018)
Y.R. Lu, M.Y. Gou, L.Y. Zhang, L. Li, T.T. Wang, C.G. Wang, Z.M. Su, Facile one-pot synthesis of hollow mesoporous fluorescent Gd2O3:Eu/calcium phosphate nanospheres for simultaneous dual-modal imaging and pH-responsive drug delivery. Dyes Pigment. 147, 514–522 (2017)
W. Xu, Y. Cui, Y.W. Hu, L.J. Zheng, Z.G. Zhang, W.W. Cao, Optical temperature sensing in Er3+-Yb3+ codoped CaWO4 and the laser induced heating effect on the luminescence intensity saturation. J. Alloys Compd. 726, 547–555 (2017)
Y.Z. Chen, F. Peng, Q.L. Zhang, W.P. Liu, R.Q. Dou, S.J. Ding, J.Q. Luo, D.L. Sun, G.H. Sun, X.F. Wang, Growth, structure and spectroscopic properties of 1 at.% Er3+: GdTaO4 laser crystal. J. Lumin. 192, 555–561 (2017)
X.J. Xue, T.L. Cheng, T. Suzuki, Y. Ohishi, KY3F10:Er3+/Yb3+ nanocrystals doped laser-induced self-written waveguide for optical amplification in the C-band, in Optical Components and Materials Xiii, ed. by S. Jiang, M.J.F. Digonnet (Spie-Int Soc Optical Engineering, Bellingham, 2016)
V.O. Kharchenko, D.O. Kharchenko, V.V. Yanovsky, Nano-sized adsorbate structure formation in anisotropic multilayer system. Nanoscale Res. Lett. 12, 337 (2017)
P.Q. Yang, J.M. Peng, Z.Y. Chu, D.F. Jiang, W.Q. Jin, Facile synthesis of Prussian blue nanocubes/silver nanowires network as a water-based ink for the direct screen-printed flexible biosensor chips. Biosens. Bioelectron. 92, 709–717 (2017)
L.I. Fockaert, P. Taheri, S.T. Abrahami, B. Boelen, H. Terryn, J.M.C. Mol, Zirconium-based conversion film formation on zinc, aluminium and magnesium oxides and their interactions with functionalized molecules. Appl. Surf. Sci. 423, 817–828 (2017)
H.Y. Lu, H.Y. Hao, H.M. Zhu, G. Shi, Q.D. Fan, Y.L. Song, Y.X. Wang, X.R. Zhang, Enhancing temperature sensing behavior of NaLuF4:Yb3+/Er3+ via incorporation of Mn2+ ions resulting in a closed energy transfer. J. Alloys Compd. 728, 971–975 (2017)
V. Kumar, G. Bullis, G.F. Wang, Investigation of NIR-tO–REd upconversion luminescence mechanism in Y2O3:Er3+, Yb3+ and the effect of co-doping Zn in the matrix. J. Lumin. 192, 982–989 (2017)
P. Deshmukh, S. Satapathy, M.K. Singh, M.P. Kamath, A.K. Karnal, Effect of Er and Dy on IR-visible up-conversion luminescence properties of (Er0.01Dy0.01La0.01Zr0.02Y0.95)2O3 transparent ceramic. Ceram. Int. 43, 14257–14262 (2017)
E. Ballem, A. Azeem, P.R. Rayavarapu, H. Divi, Structural and luminescent studies of erbium-doped CaZrO3 green-emitting nanophosphors. Luminescence 32, 1246–1251 (2017)
B. Sikora, P. Kowalik, J. Mikulski, K. Fronc, I. Kaminska, M. Szewczyk, A. Konopka, K. Zajdel, R. Minikayev, K. Sobczak, W. Zaleszczyk, A. Borodziuk, J. Rybusinski, J. Szczytko, A. Sienkiewicz, T. Wojciechowski, P. Stepien, M. Frontczak-Baniewicz, M. Lapinski, G. Wilczynski, W. Paszkowicz, A. Twardowski, D. Elbaum, Mammalian cell defence mechanisms against the cytotoxicity of NaYF4:(Er,Yb,Gd) nanoparticles. Nanoscale 9, 14259–14271 (2017)
B. Xu, P. Wang, X. Meng, K. Zou, J. Liu, Effects of the Ho3+/Yb3+ concentration ratio on the structure and photoluminescence of ZnO films. J. Lumin. 175, 78–81 (2016)
J.C. Li, S.G. Li, H.F. Hu, F.X. Gan, Emission properties of Yb3+/Er3+ doped TeO2-WO3-ZnO glasses for broadband optical amplifiers. J. Mater. Sci. Technol. 20, 139–142 (2004)
X.Y. Zhang, H. Yu, L.Y. Guo, J.N. Jin, Q.X. Li, Y.A. Guo, Y.T. Fu, Y.H. Shi, L.J. Zhao, Comprehensive model and investigation of F– ions-induced cubic-to-hexagonal phase transformation in NaYF4. J. Alloys Compd. 728, 1254–1259 (2017)
A.J. Huang, Z.W. Yang, C.Y. Yu, J.B. Qiu, Z.G. Song, Splitting upconversion emission and phonon-assisted population inversion of Ba2Y(BO3)2Cl: Yb3+, Er3+ phosphor. J. Am. Ceram. Soc. 100, 4994–4998 (2017)
W.W. Liu, H.Y. Zhang, H.G. Wang, M. Zhang, M. Guo, Titanium mesh supported TiO2 nanowire arrays/upconversion luminescence Er3+-Yb3+ codoped TiO2 nanoparticles novel composites for flexible dye-sensitized solar cells. Appl. Surf. Sci. 422, 304–315 (2017)
H. Akazawa, H. Shinojima, Comparative study of visible and infrared photoluminescence resulting from indirect and direct excitation processes of Er3+ ions doped in ZnO host films. J. Appl. Phys. 122, 10 (2017)
L. Xu, J.W. Zhang, H. Zhao, H.B. Sun, C.X. Xu, Enhanced photoluminescence intensity by modifying the surface nanostructure of Nd3+-doped (Pb, La)(Zr, Ti)O3 ceramics. Opt. Lett. 42, 3303–3306 (2017)
S. Khan, H. Choi, S.Y. Lee, K.R. Lee, O.M. Ntwaeaborwa, S. Kim, S.H. Cho, Unexpected roles of interstitially doped lithium in blue and green light emitting Y2O3:Bi3+: a combined experimental and computational study. Inorg. Chem. 56, 12139–12147 (2017)
P.P. Sukul, M.K. Mahata, K. Kumar, NIR optimized dual mode photoluminescence in Nd doped Y2O3 ceramic phosphor. J. Lumin. 185, 92–98 (2017)
F. Auzel, Application des transferts d’énergie résonnants a l’effet laser de verres dopés avec Er3+. Ann. Telecommun. 24, 363–376 (1969)
V.V. Ovsyankin, P.P. Feofilov, Mechanism of summation of electronic excitations in activated crystals. Sov. J. Exp. Theor. Phys. Lett. 3, 322 (1966)
V.E. Karapetyan, B.I. Maksakov, P.P. Feofilov, Absorption and luminescence of divalent samarium in alkali-halide single crystals. Opt. Spectry 14, 441–443 (1963)
P.V. Ramakrishna, T.L. Rao, A. Singh, B. Benarji, S. Dash, Structural and photoluminescence behavior of thermally stable Eu3+ activated CaWO4 nanophosphors via Li+ incorporation. J. Mol. Struct. 1149, 426–431 (2017)
Y.L. Yan, W. Zhang, B. Ren, L.S. Zhong, Y.H. Xu, Effects of Li+ co-doping on the concentration quenching threshold and luminescence of GdVO4:Eu3+ nanophosphors. Ionics 23, 869–875 (2017)
X.W. Wang, X. Zhang, Y.B. Wang, H.Y. Li, J. Xie, T. Wei, Q.W. Huang, X.J. Xie, L. Huang, W. Huang, Comprehensive studies of the Li+ effect on NaYF4: Yb/Er nanocrystals: morphology, structure, and upconversion luminescence. Dalton Trans. 46, 8968–8974 (2017)
Y.B. Wang, T. Wei, X.W. Cheng, H. Ma, Y. Pan, J. Xie, H.Q. Su, X.J. Xie, L. Huang, W. Huang, Insights into Li+-induced morphology evolution and upconversion luminescence enhancement of KSc2F7:Yb/Er nanocrystals. J. Mater. Chem. C 5, 6 (2017)
A.N. Meza-Rocha, B.F. Huerta, U. Caldino, S. Carmona-Tellez, M. Bettinelli, A. Speghini, S. Pelli, G.C. Righini, C. Falcony, Dependence of the up-conversion emission of Li+ co-doped Y2O3:Er3+ films with dopant concentration. J. Lumin. 167, 352–359 (2015)
D. Avram, B. Cojocaru, I. Tiseanu, M. Florea, C. Tiseanu, Down-/Up-conversion emission enhancement by li addition: improved crystallization or local structure distortion? J. Phys. Chem. C 121, 14274–14284 (2017)
A.L. Patterson, The scherrer formula for X-ray particle size determination. Phys. Rev. 56, 978–982 (1939)
D. Doutt, H.L. Mosbacker, G. Cantwell, J. Zhang, J.J. Song, L.J. Brillson, Impact of near-surface defects and morphology on ZnO luminescence. Appl. Phys. Lett. 94, 252103 (2009)
Q. Zhu, J.G. Li, X. Li, X. Sun, Morphology-dependent crystallization and luminescence behavior of (Y, Eu)2O3 red phosphors. Acta Mater. 57, 5975–5985 (2009)
H. Song, J. Wang, B. Chen, H. Peng, S. Lu, Size-dependent electronic transition rates in cubic nanocrystalline europium doped yttria. Chem. Phys. Lett. 376, 1–5 (2003)
M. Liu, S.W. Wang, D.Y. Tang, L.D. Chen, J. Ma, Preparation and upconversion luminescence of YAG(Y3Al5O12):Yb3+, Ho3+ nanocrystals. J. Rare Earth. 27, 66–70 (2009)
G.Y. Chen, H.C. Liu, G. Somesfalean, Y.Q. Sheng, H.J. Liang, Z.G. Zhang, Q. Sun, F.P. Wang, Enhancement of the upconversion radiation in Y2O3Er3+ nanocrystals by codoping with Li+ ions. Appl. Phys. Lett. 92, 3 (2008)
L.D. Sun, C. Qian, C.S. Liao, X.L. Wang, C.H. Yan, Luminescent proper-ties of Li+ doped nanosized Y2O3: Eu. Solid State Commun. 119, 393–396 (2001)
A. Pandey, V.K. Rai, K. Kumar, Influence of Li+ codoping on visible emission of Y2O3: Tb3+, Yb3+ phosphor, Spectrochim. Acta A Mol. Biomol. Spectrosc. 118, 619–623 (2014)
T. Fan, Q.Y. Zhang, Z.H. Jiang, The effects of Li on the near-infrared luminescence properties of Nd3+/Li+ codoped Y2O3 nanocrystals. Opt. Commun. 284, 1594–1597 (2011)
Y.F. Bai, Y.X. Wang, G.Y. Peng, K. Yang, X.R. Zhang, Y.L. Song, Enhance upconversion photoluminescence intensity by doping Li+ in Ho3+ and Yb3+ codoped Y2O3 nanocrystals. J. Alloys Compd. 478, 676–678 (2009)
K. Mishra, S.K. Singh, A.K. Singh, S.B. Rai, Frequency upconversion in Er3+ doped Y2O3 nanophosphor: Yb3+ sensitization and tailoring effect of Li+ ion. Mater. Res. Bull. 48, 4307–4313 (2013)
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51352002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, B., Liu, J. & Zou, K. The photo-switch effect and the energy-level population change of Li+ doping in Yb3+/Er3+ co-doped Y2O3 upconversion films. Appl. Phys. A 125, 100 (2019). https://doi.org/10.1007/s00339-018-2371-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-018-2371-1