
Abstract
We study experimentally the electronic excitation mechanisms involved in the breakdown and ablation of wide band gap dielectrics. A femtosecond pump–probe interferometry technique, with 100 fs temporal resolution, allows measuring the modification of refractive index induced by ultra-short intense laser pulses. To get more information in the complex process of excitation and relaxation mechanisms involved during and after the interaction, we use a sequence of two excitation pulses: a first short pulse at 400 nm excites a controlled density of carriers, and a second one at 800 nm with variable pulse duration, from 50 fs to 10 ps, reaches an excited solid. In Al2O3, we show that the total density of carriers never exceeds the sum of the densities excited by the two pulses sent independently. This means that the second pulse deposits further energy in the material by heating the previously excited carriers, and that no electronic multiplication occurs. On the other hand, in SiO2, it is possible, under specific conditions, to observe an increase of carrier density due to impact ionization. All these results demonstrate that the avalanche process, which is often invoked in the laser breakdown literature, does not play a dominant role in optical breakdown induced by short pulses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Despite a large number of published works during the last fifteen years, the physical mechanism leading to the optical breakdown of wide band gap materials by ultra-short laser pulses still remains unclear.
This is probably because the majority of experiments are concentrated on ablation threshold measurements [1–3]. However, this process is complex, involving a complicated temporal evolution of the sample and the laser pulse, both being linked by the change of optical constants of the solid during the pulse. The latter is induced by the promotion of a large density of carriers, electrons in the conduction band and holes in the valence band. If one concentrates only on this initial process of electronic excitation, the controversy still exists, and the importance of different possible excitation mechanisms is still under debate [3–9]. For transparent dielectrics, the photon energy of femtosecond lasers is usually much below the band gap, and the dominant initial excitation process must be nonlinear: multiphoton or tunnelling ionisation.
Then the excited carriers can absorb a large number of photons within the conduction band [5] and, if their energy is high enough, collide with valence band electrons. The iteration of this process is the avalanche ionisation. In the recent literature, one can find advocates of all these three different mechanisms: multiphoton [6], avalanche [7, 8] or tunnelling [3, 9]. Theoretical models have been based on either simplified rate equations [10] or a kinetic approach of Boltzmann’s equations [11, 12]. The purpose of the present work is to present experimental measurements of the density of carriers achieved in a dielectric just after interaction with one or two laser pulses. More precisely, the aim of our time-resolved pump–probe studies is to clearly identify the excitation mechanism at work, by using two excitation (pump) pulses, and separating the different excitation steps: excitation from the valence band to the conduction band, and further excitation or heating of the carriers in the conduction band.
2 Time-resolved interferometry: principle and experimental set-up
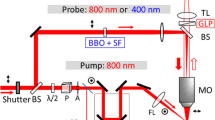
The experiments have been made at the Saclay Laser–Matter Interaction Center (SLIC: http://iramis.cea.fr/slic/index.php) facility. The laser is a CPA amplified Ti–Sa system, delivering up to 70 mJ at 800 nm, with a repetition rate of 20 Hz. The schematic drawing of the experimental set-up is displayed in Fig. 1. The beam is initially separated into two beam lines each equipped with a compressor. We use one beam line to generate the short pump and the probe pulses, and the other beam line for the second pump with variable duration. Then one of the short IR pulses is converted into the second harmonic in a thin BBO crystal. In this way three beams necessary for the double-pump experiment are obtained: the short UV beam as the first pump, the short IR beam used then as the probe and the IR beam with variable pulse duration for the second pump. The last two pulses could be independently delayed using micron-precision motorised stages. We are focusing both pump beams on the surface of the sample (crystalline cubes 5×5×5 mm3, or wafers 10×10×0.5 mm3) to avoid propagation effects. Spatial overlapping of those two beams is ensured by illuminating the surface with a green laser diode and imaging with a CCD camera.

Diagram of the experimental set-up for pump–probe interferometry with one or two pump pulses: one of the pumps is the second harmonic at 400 nm, with pulse duration 50 fs, and the other is the fundamental at 800 nm, with variable pulse duration. The two probe pulses are generated in a Michelson interferometer and are tilted in space
The probe system consists of two identical pulses that are created in a Michelson interferometer after going through the sample. It is essential hereby to recall that the pump beam diameter is several (ten to four) times smaller than the probe and reference beam diameter on the surface. Thus, one pulse (reference) is translated in the plane of incidence, so that the excited, central part of the probe beam overlaps with the unexcited, peripheral part of the translated reference beam. Finally, they will interfere at the exit of a monochromator, which acts as a stretcher, ensuring overlapping in time of the two pulses. This interference image is shown in Fig. 2: the horizontal axis is the wavelength, and the vertical axis is the vertical axis in real space, along the entrance slit. The phase shift experienced by the central part of the probe beam which crossed the excited part of the sample is clearly visible in this figure (in the centre).

Interferometry image observed at the exit of the monochromator. The central part of the probe beam, which has crossed a perturbed (excited by the pump pulse) region of the sample, experiences a phase shift clearly visible in the picture. Note also the modification of the contrast, indicating that part of the second probe pulse has been absorbed
The experiment consists in recording this interference image for each pump–probe delay, and to perform a Fourier analysis to extract the phase and amplitude of each horizontal line. From this, we get the phase shift and absorption. To avoid any cumulative effect, the sample is moved after each shot.
The phase shift is proportional to the variation of the real part of the refractive index Δn of the solid: Δϕ=2πΔnL/λ, where L is the length over which the pump and probe beams overlap, and λ is the probe wavelength.
We have developed a simple model, valid upon moderate excitation, to calculate the change of dielectric function induced by the presence of excited carriers, based on the Drude model. The complete calculation is explained in a previous paper [13], but for the sake of simplicity we can assume that there are three contributions depending on the material, and we can write Δn as
In this equation, the first term corresponds to the Kerr effect, n 2 being the nonlinear refractive index and I p the pump beam intensity. The second term is always negative and it stands for the free-carrier generation. It is proportional to the excitation density in the medium N CB. The third term illustrates the appearance of levels in the band gap due to the formation of self-trapped excitons and it is proportional to the density of these trapped carriers N tr. Finally, e is the elementary charge, n 0 is the unperturbed refractive index of the sample, ε 0 is the vacuum permittivity, m ∗ is the electron or hole effective mass, ω is the probe pulsation and ω tr is the energy difference between the fundamental and the first excited state of the induced defect.
When we measure the evolution of the change of the phase shift—which again is proportional to the change of refractive index—we observe different behaviours in SiO2 (Fig. 3, left) and Al2O3 (Fig. 3, right). In both cases, the positive phase shift due to the Kerr effect is followed by an abrupt change towards a negative value due to the promotion of carriers in the conduction band (negative contribution, second term of Eq. (1)). Finally, the evolution of the density of free carriers can be followed in time. Whereas it is stable at this time scale of 2 ps in Al2O3, it can be seen that the trapping of electron–hole pairs due to the formation of self-trapped excitons is extremely fast in SiO2, leading to a final positive phase shift. The self-trapping rate is 150 fs and, after about 700 fs, all carriers have been trapped in the band gap.

Phase shift as a function of time measured in SiO2 (left) and Al2O3 (right). The initial positive phase shift is due to the Kerr effect, then free carriers induce a negative phase shift (plasma effect) and finally in the case of SiO2 electron–hole pairs are trapping deeply in the band gap, leading to a positive phase shift. The intensities are kept well below the threshold for both cases
In a previous set of experiments, we have studied the evolution of the carrier density, as a function of laser intensity, below and above breakdown threshold, for different pulse durations and in different materials. This is achieved by measuring the negative phase shift observed in Fig. 3 at a fixed time delay, as a function of the pump laser intensity. For short pulses (60 fs), the phase-shift evolution measured in Al2O3 is shown in Fig. 4, and our conclusions were the following [14]:
-
First, at low intensity, i.e. a few TW/cm2, the density of carriers increases nonlinearly, following a multiphoton excitation rule: the phase shift measured by the interferometry technique is proportional to I N, where I is the laser intensity and N the minimal number of photons needed to promote an electron from the valence band to the conduction band. This is the signature of a multiphoton excitation.
-
Then, when we approach the breakdown threshold, this highly nonlinear evolution saturates. We have shown that this saturation can be explained if one takes into account the evolution of the optical properties of the solid. Since the critical density is reached during the pulse, the surface of the sample becomes highly reflective, and a thin layer of dense plasma behind the surface strongly absorbs the remaining part of the pulse. These two effects combine to prevent further penetration of the pump beam, and thus the saturation of the signal. It should be noted that this result has been confirmed later by another group which has performed similar measurements on SiO2 and Al2O3, and has arrived at the same conclusions [6].
Furthermore, using the data obtained from our interferometry experiments, we could develop a simple model, which describes both the propagation of the pump pulse and the evolution of the solid. With this model we have been able to simulate the evolution of the craters’ depth as a function of the laser intensity, and obtain a good quantitative agreement with the measured craters [15].

Phase shift as a function of laser intensity measured in Al2O3. The pump wavelength is 800 nm and the duration is 60 fs. The line is indicating an I 6 dependence law. The arrow and empty circle indicate the breakdown threshold
At that point we could think that the process of electronic excitation is entirely understood. However, when we reach the breakdown intensity, the excitation process may change and this can be obscured for two reasons. First, this change of excitation mechanism could occur only in a very thin layer at the surface, which contributes to only a part of the signal and cannot be detected. Second, such an effect may also be hidden anyway by the saturation process, which is unavoidable since the critical density is attained at the surface. Thus, to complete our experimental data and get a more detailed knowledge of the excitation and energy deposition processes, we decided to carry out double-excitation measurements.
3 Double-pump experiments: electron heating mechanism
The idea of the double-pump experiment [16] is to try to observe the different stages of the electronic excitation: from the valence band to the conduction band, and then inside the conduction band. We already know from photoemission measurements that the energy of electrons heated by a short laser pulse can exceed the energy of incident photons by more than one order of magnitude [17]. On the other hand, the process of impact ionisation, which may give rise to avalanche ionisation, should result in an increase of the excitation density. Thus, the combination of two pump pulses should allow the observation of the impact ionisation seeded electrons, if any.
The principle of the double-pulse experiment is the following: we first excite a large density of carriers (1020 cm−3 [18]) with a short ultraviolet (400 nm) pulse. Following this excitation, a fast relaxation (tenths of fs) of the electron energy takes place in the conduction band. At this stage, and before any electron–hole recombination or trapping starts taking effect, we send a second pulse, at the fundamental wavelength (800 nm), to re-excite the solid. We expect the latter to heat the carriers sufficiently in the conduction band to seed the impact ionisation process if it would occur. The delay between the two pump pulses is chosen in such a way that the infrared pulse will ‘see’ a maximal excitation density and that there is no overlap in time between the two pulses to avoid any crossed-excitation effect.
Finally, we measure the evolution of the carrier density as a function of time, at all probe delays from before the first pump pulse until after the second pump pulse. The intensity of each pulse is kept below the damage threshold (at least under 70 % of it), such that the breakdown occurs only when the sequence of the two pulses is applied. The damage threshold is being measured experimentally by monitoring any phase change on the surface of the sample, on a screen connected to a CCD camera.
We have chosen to investigate the two above-mentioned materials, since we expect different behaviours owing to the free carrier relaxation dynamics that differ strongly, having a long (several hundreds of ps) lifetime in the conduction band (Al2O3) and forming self-trapped excitons in a short time scale (150 fs trapping rate) in SiO2.
3.1 Double-pulse experiments: Al2O3
In Fig. 5 is presented the measured phase shift with only the second pulse (the fundamental) and both pulses. In this case both pulses are short, and the delay between them is 5 ps. The UV pulse energy is 2.6 μJ (11 μm waist, 0.48 mm Rayleigh length) and the IR energy is 27 μJ (20 μm waist, 1.57 mm Rayleigh length). The probe’s waist is 190 μm and its Rayleigh length is 41 mm. Both energies are well below the single-shot damage threshold. Only when both pump pulses are combined is the damage threshold reached. The spatial overlapping length L is ∼41 μm. This is being achieved by using a perpendicular configuration between the pump and the probe pulses. In this case, the probe is being reflected by the surface of the sample by internal reflection.

Phase shift as a function of time with one pump at 800 nm (circles) and two pump (400 nm and 800 nm 5 ps later, both 50 fs duration, see text) pulses (full circles) measured in α-Al2O3. Fluences are 0.68 J/cm2 for UV and 2.15 J/cm2 for IR
The measurement has been made first with the second pulse alone (open circles in Fig. 5). One observes a small positive phase shift at delay 5.5 ps, due to the Kerr effect. The Kerr effect induces a nonlinear polarisation which gives rise to a positive change of refractive index, and lasts only while the pump and probe pulses overlap in time. Then the phase shift becomes negative, due to the presence of free electron–hole pairs in the solid. This negative signal evolves slowly, following the decrease of the density of carriers. In the second experiment (full triangles), we send a pulse at 400 nm before this IR pulse to generate an initial excitation density. When the second pulse arrives, it will further excite the solid, and in this case we do reach the breakdown threshold. The signal due to excited carriers increases after this second pulse, but the important result is that the total signal (phase shift of −0.67) is equal, within error bars, to the sum of the two signals measured with independent pulses: phase shift of −0.27 rad due to the first pulse, plus phase shift of −0.42 rad due to the second pulse. In other words, the density of carriers excited by the sequence of two pulses is equal to the addition of the ones obtained with each pulse separately. This seems to indicate that the process of electronic avalanche, which would have increased the density of carriers, does not lead to a measurable effect in this case.
After this first experiment our aim was to test a different experimental condition and change the duration of the second pulse. Indeed, avalanche ionisation is a sequential process: excitation of carriers from the valence band into the conduction band, followed by a cycle of excitation within the conduction band and impact ionisation. Thus, it may take some time for the avalanche process to develop, and this has been shown in a theoretical paper [12]. Thus, our idea was that it may be possible to observe avalanche if one increases the duration of the second pump pulse. The result of the measurement is shown in Fig. 6. We have measured the complete decay kinetics with a short (50 fs, 7.3 μJ) pump pulse at 400 nm alone, and also with the long pump pulse (10 ps, 130 μJ) at 800 nm alone. With the second harmonic we create a large excitation density, while no change of the refractive index is measured with the long IR pulse. This is because the peak intensity, which is equal to 1×1012 W/cm2 for this second excitation pulse, is too low to generate efficiently any free electrons by a nonlinear process. However, when the two pulses are combined (with a 5 ps delay at intensity peak) we observe, apart from reaching the damage threshold, a change in the time evolution of the negative phase shift. But, instead of an increase of the signal, as we would have expected if an avalanche had occurred, we observe a decrease of the signal, or more precisely an acceleration of the decrease of the signal. Since this negative phase shift is proportional to the density of free carriers, this means that the arrival of the second pump pulse induces an acceleration of the recombination process, or that a new positive contribution to the change of refractive index appears.

Phase shift as a function of time with one pump at 400 nm (full circles) and two pump (400 nm, 50 fs, and 800 nm 5 ps later, 10 ps duration, see text) pulses (triangles) measured in α-Al2O3. Fluences are 2 J/cm2 for UV and 10.34 J/cm2 for IR
If we suppose that the electron–hole recombination dynamics can be described by a bimolecular recombination model, it should follow the following equation:
where N is the electron or hole density, σ is the electron hole capture cross section and v the velocity of carriers in the solid. This gives the following evolution for the excited carrier density:
In the framework of this model, a speeding up of the recombination kinetics can be explained if either N 0, the initial density, or the product σv, which gives the electron– hole recombination probability per unit time, is increased. Our measurement shows that the initial density is unchanged, since we do not see any increase of the phase shift during the second pump pulse. This means that either the electron–hole capture cross section or the carrier velocity is increased. When the second pump pulse interacts with the solid, it will induce a heating of the free carriers by multiphoton or sequential single-photon absorption. Thus, their average kinetic energy will increase. We do not expect on the contrary an increase of the cross section, which is usually maximum when the kinetic energy tends to zero. Thus, we believe that the faster decay kinetics for the measured phase shift is a signature of the carrier heating by the second pulse.
Finally, what is the mechanism at work in this double-pulse experiment? When we send the second pump pulse, we reach the breakdown threshold. However, the total number of electrons does not change. On the contrary, we observe a faster decrease of the negative phase shift, which indicates a faster electron–hole pair recombination. This acceleration can be due to the increase of the average kinetic energy of free carriers in the solid.
An alternative explanation for the change of phase shift that follows the interaction with the second pulse could be an increase of temperature, which indeed should contribute positively to the change of the refractive index, since the latter is known to increase when the temperature increases [19].
In both hypotheses, we can conclude from this observation that the breakdown is due to the injection of further energy in the solid, by single or multiphoton excitation of the electrons (respectively holes) in the conduction band (respectively valence band). Thus, no avalanche can be observed in our measurement, but the damage of the solid is occurring because a critical amount of energy is absorbed, and this energy is transferred from the second pump pulse to the carriers excited by the first pulse.
3.2 Double-pulse experiments: SiO2
In this second part, we continue our investigation on quartz. In Fig. 7 is plotted the phase shift for the two pump beams alone and for the combination of those two beams in a train of pulses. The delay between the two pulses is 200 fs. The second-harmonic pulse (13.5 μm waist, 1.4 mm Rayleigh length) arrives first at a fluence of 1.17 J/cm2 (50 fs pulse duration) without damaging the sample. Later, the fundamental pulse (56.6 μm waist, 12.6 mm Rayleigh length) reaches the surface of the sample at a fluence of 1.66 J/cm2 (300 fs pulse duration); this intensity corresponds to 17 % of the IR single-shot threshold intensity. The overlapping length L, in this configuration (quasi-co-linear, see Fig. 1), is 164 μm.

Phase shift as a function of time with one pump at 400 nm (squares), one pump at 800 nm (reversed triangles) and two pump (400 nm, 50 fs, and 800 nm 200 fs later, 300 fs duration, see text) pulses (full circles) measured in SiO2. Fluences are 1.17 J/cm2 for UV and 1.66 J/cm2 for IR
First, one can observe a positive phase shift, which corresponds to the Kerr effect, followed by a negative signal, a signature of free-carrier generation and finally a positive phase shift that is attributed to the formation of self-trapped excitons. Now, although the fundamental pulse (reversed triangles) is not exciting any carriers on its own, we can clearly see a rise in the absolute value of the negative phase shift (∼50 % rise) when both beams are present (full circles curve) and while being above the damage threshold for the sequence of the two pulses.
This suggests that the 800-nm beam is ‘heating’ the free carriers previously excited by the second harmonic. Then the hot carriers which have reached a high enough energy are able to induce impact ionisation, thus increasing the final density. One can also observe the rise in the final positive phase shift—proportional to the excitation density—that confirms the multiplication of the electrons suggested above. Furthermore, two parameters have been investigated in depth: fundamental pulse duration and delay between pump pulses. This has revealed that impact ionisation is more efficient for the set of parameters listed above. In particular, the pulse of 300 fs duration corresponds to the ‘free’ electron lifetime in the conduction band and the delay between the pulses is fixed so that the second pulses will ‘see’ a maximum excitation density.
4 Conclusion and perspective
We believe that our experimental investigation of the excitation density reached in single- and double-pulse excitation brings new and valuable information about the electronic excitation processes occurring during the breakdown of dielectric materials induced by short laser pulses. The first pulse excites free carriers, and the second pulse will give further energy to these pre-excited carriers, thus overcoming the damage threshold. Damaging is taking place when a critical amount of energy has been deposited in the solid, inducing fusion/sublimation of the solid, and the damage threshold does not correspond to any critical excitation density. In materials with long free electron lifetime, there is no reason why the sequence of events would be different in the case of damaging by a single laser pulse: the roles of the first and second pulses will be played by the leading edge and the tail of the pulse, respectively. Comparison is more subtle in the case of materials where trapping occurs. Modelling and simulating this interaction will give access to parameters such as the absorption cross sections for electrons in the conduction band, thus being able to simulate the interaction in the case of one short pulse and accessing the efficiency of different ionization channels.
In conclusion, the principal excitation mechanism is revealed to be the multiphoton absorption, but the level of dominance of the latter is dependent on the nature of the material. Further experimental investigation should include longer probe temporal delays to follow up the final state after recombination and/or trapping of the electrons. In that case, adjusting longer delays between the two pump pulses can be an interesting parameter to explore in order to reveal the material’s behaviour, interacting with a sequence of pulses, at later times.
References
B.C. Stuart, M.D. Feit, S. Herman, A.M. Rubenchik, B.W. Shore, M.D. Perry, Phys. Rev. B 53, 1749 (1996)
M. Lenzner, J. Krüger, S. Sartania, Z. Cheng, Ch. Spielmann, G. Mourou, W. Kautek, F. Krausz, Phys. Rev. Lett. 80, 4076 (1998)
B. Chimier, O. Uteza, N. Sanner, M. Sentis, T. Itina, P. Lassonde, F. Légaré, F. Vidal, J.C. Kieffer, Phys. Rev. B 84, 094104 (2011)
D. Grojo, M. Gertsvolf, S. Lei, T. Barillot, D.M. Rayner, P.B. Corkum, Phys. Rev. B 81, 212301 (2010)
D. Arnold, E. Cartier, Phys. Rev. B 46, 23 (1992)
V. Temnov, K. Sokolowski-Tinten, P. Zhou, A. El-Khamhawy, D. Von Der Linde, Phys. Rev. Lett. 97, 237403 (2006)
L. Englert, B. Rethfeld, L. Haag, M. Wollenhaupt, C. Sarpe-Tudoran, T. Baumert, Opt. Express 15, 17855 (2007)
P.P. Rajeev, M. Gertsvolf, P.B. Corkum, D.M. Rayner, Phys. Rev. Lett. 102, 083001 (2009)
I.N. Zavestovskaya, P.G. Eliseev, O.N. Krokhin, N.A. Men’kova, Appl. Phys. A 92, 903 (2008)
B. Rethfeld, Phys. Rev. B 73, 035101 (2006)
P. Daguzan, P. Martin, S. Guizard, G. Petite, Phys. Rev. B 52, 17099 (1995)
A. Kaiser, B. Rethfeld, M. Vicanek, G. Simon, Phys. Rev. B 61, 11437 (2000)
Ph. Martin, S. Guizard, Ph. Daguzan, G. Petite, P. D’Oliveira, P. Meynadier, M. Perdrix, Phys. Rev. B 55, 5799 (1997)
F. Quéré, S. Guizard, P. Martin, Europhys. Lett. 56, 138 (2001)
S. Guizard, A. Semerok, J. Gaudin, M. Hashida, P. Martin, F. Quéré, Appl. Surf. Sci. 186, 364 (2002)
X.A. Shen, S.C. Jones, P. Braunlich, Phys. Rev. Lett. 62, 2711 (1989)
A. Belsky, P. Martin, H. Bachau, A.N. Vasil’ev, B.N. Yatsenko, S. Guizard, G. Geoffroy, G. Petite, Europhys. Lett. 67(2), 301 (2004)
S. Guizard, Ph. Martin, Ph. Daguzan, G. Petite, P. Audebert, J.P. Geindre, A. Dos Santos, A. Antonetti, Europhys. Lett. 29, 401 (1995)
E.S. Zouboulis, M. Grimsditch, J. Appl. Phys. 70, 772 (1991)
Acknowledgements
Ecole Polytechnique is gratefully acknowledged for offering a visiting grant to Sergey Klimentov. We also would like to acknowledge the laser team in Saclay who carried out valuable work to have the laser system operating properly every day. This research has been funded by the French National Research Agency (ANR: programs Nanomorphing and FLAG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mouskeftaras, A., Guizard, S., Fedorov, N. et al. Mechanisms of femtosecond laser ablation of dielectrics revealed by double pump–probe experiment. Appl. Phys. A 110, 709–715 (2013). https://doi.org/10.1007/s00339-012-7217-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00339-012-7217-7