
Abstract
With the increase in knowledge resulting from the sequencing of the human genome, the genetic basis for the underlying differences in individuals, their diseases, and how they respond to therapies is starting to be understood. This has formed the foundation for the era of precision medicine in many human diseases that is beginning to be implemented in the clinic, particularly in cancer. However, preclinical testing of therapeutic approaches based on individual biology will need to be validated in animal models prior to translation into patients. Although animal models, particularly murine models, have provided significant information on the basic biology underlying immune responses in various diseases and the response to therapy, murine and human immune systems differ markedly. These fundamental differences may be the underlying reason why many of the positive therapeutic responses observed in mice have not translated directly into the clinic. There is a critical need for preclinical animal models in which human immune responses can be investigated. For this, many investigators are using humanized mice, i.e., immunodeficient mice engrafted with functional human cells, tissues, and immune systems. We will briefly review the history of humanized mice, the remaining limitations, approaches to overcome them and how humanized mouse models are being used as a preclinical bridge in precision medicine for evaluation of human therapies prior to their implementation in the clinic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Precision medicine as defined by NIH is “an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person. This approach will allow clinicians and researchers to predict more accurately precise treatments and prevention strategies for a particular disease in individual patients” (https://ghr.nlm.nih.gov/primer/precisionmedicine/definition). An example of precision medicine that has been followed for many years is how bacterial infections are treated. Diagnosis of a bacterial infection is commonly followed by exposure of the pathogen to a number of antibiotics known to be effective against that bacterial species. The antibiotic(s) that is most effective against the bacteria in the lab tests is prescribed to the patient, making this a “precision medicine” approach based on knowledge of the bacteria and the mechanism of action of the antibiotic. However, this could also be considered “personalized medicine”. These two terms have significant overlap and are often used interchangeably. We will use the term precision medicine for the variability of human immune responses in disease and in response to therapies.
The ability to fully utilize precision medicine is limited by logistical and ethical restrictions of performing experiments in humans, particularly in children (Davis 2012). Any therapies or treatments that are being considered for “precision medicine” need to be tested in preclinical models prior to their translation into the clinic. For this, many investigators have turned to animal models as surrogates of human immunology, each with its advantages and disadvantages (Perrin 2014). Mice are the most widely used animal models for the study of immunology and have provided many fundamental insights into how mammalian immune systems develop and the interactive function of the many immune cell populations (Rosenthal and Brown 2007). Observations in mice, however, often do not directly translate into the clinic (Warren et al. 2014). This is particularly striking in murine models of cancer. For example, there are multiple murine models available for the study of cancer, and fundamental information about tumors and immune system interactions have been discovered. However, as recently reviewed (Landgraf et al. 2018) “…murine cancer models often still do not recapitulate basic tumour biology, tumour-microenvironment interactions, drug responses and therapy resistance when compared with human disease (Ellis and Reardon 2009).” The “disconnect” between therapies that are effective in mice but not in humans is likely due to multiple differences in murine and human genomes, molecular and metabolic pathways, and in immune systems, particularly in their innate immune systems (DeBerardinis and Chandel 2016; Hagai et al. 2018; Kawai and Akira 2010; Mestas and Hughes 2004; Pavlova and Thompson 2016; Rosenthal and Brown 2007; Seok et al. 2013; Takao et al. 2015; Takao and Miyakawa 2015; Warren et al. 2014).
Mice and humans display multiple discrepancies in their genetics and in their immune systems, suggesting a possible fundamental reason for the lack of direct translation of some findings in mice to patients, particularly with respect to innate immune responses (Hagai et al. 2018; Mestas and Hughes 2004; Sato et al. 2014; Seok et al. 2013; Takao et al. 2014, 2015; Takao and Miyakawa 2014, 2015; Warren et al. 2014). An example is that human endothelial cells can express MHC class II, a molecule important in antigen presentation, whereas murine endothelial cells do not, providing a unique source of antigen-presenting cells (APC) in humans not present in mice (Choo et al. 1997). In another example, genomic mouse inflammatory responses do not mimic the genomic responses observed during human inflammation (Takao and Miyakawa 2015), although this has been debated in the literature (Seok et al. 2013; Takao et al. 2015; Warren et al. 2014). The data analyses of both groups, however, show that there is only a 5.9–15.0% overlap between these genomic responses. Furthermore, there are many differences in expression of toll-like receptors (TLRs) on innate immune cells. Although TLR1-TLR9 are identical, TLR 10 is not active in mice and TLR11, TLR12, and TLR13 have been lost from the human genome (Kawai and Akira 2010), suggesting fundamental differences in the molecular components of innate immunity. In addition, many of the immune-modulating drugs entering the clinic are human-specific, including monoclonal antibodies (mAbs) that cannot be evaluated in standard mouse models due to species specificity (Donini et al. 2018; Klausen et al. 2019; Lee et al. 2018; Wilkinson and Leishman 2018).
An emerging technology that addresses the species specificity of the human immune system and human biology is the creation of immunodeficient mice engrafted with functional human immune systems, i.e., “humanized mice”. Humanized mice permit the investigation of functional human cells and tissues and allow potential therapies to be tested for safety and efficacy without putting patients at risk prior to the introduction of new therapeutics into the clinic.
Brief history of humanized mice
Humanized mice have become important pre-clinical tools due to improvements over the last 30 years in the immunodeficient mice used for human cell and tissue engraftment, particularly in the engraftment of a functional human immune system (Shultz et al. 2012, 2007). The immunodeficient CB.17-Prkdcscid (scid) strain of mice described in 1983 (Bosma et al. 1983) was used in 1988 to engraft human hematopoietic stem cells (HSCs) (Kamel-Reid and Dick 1988; McCune et al. 1988) and human peripheral blood mononuclear cells (PBMCs) (Mosier et al. 1988) The CB.17-scid strain has a defect in the Prkdc gene, which encodes the catalytic subunit of the DNA-dependent protein kinase, leading to defects in DNA repair, preventing the recombination of T cell and B cell receptors and a loss of adaptive immunity (Blunt et al. 1996). However, engraftment with human hematopoietic and immune cells was extremely low in CB.17-scid mice, limiting its utility for the study of human immune systems.
In the mid 1990s, the NOD-scid strain was described (Koyanagi et al. 1997; Shultz et al. 1995), and this strain showed enhanced engraftment of human HSC, PBMC and a variety of human cancers, particularly hematopoietic cancers (Greiner and Shultz 1998; Shultz et al. 2007). The improved engraftment was thought to be due to the relative deficiencies in host innate immunity, particularly defects in NK cells whose function hindered human hematopoietic and immune cell engraftment (Shultz et al. 1995). It was later discovered that a key component of the NOD strain, in addition to its depressed innate immune system and NK cell defects, was its novel allele at the signal regulatory protein alpha (Sirp-a) locus. Sirp-α is expressed by cells of the myeloid lineage including macrophages (Barclay and Brown 2006), and binds to CD47, which is expressed on most hematopoietic as well as non-hematopoietic cells (Brown and Frazier 2001). Sirp-α/CD47 interaction provides a “do not eat me” signal to the macrophage (Barclay and Brown 2006). NOD strain mice have a Sirp-α polymorphism that is very similar to that of humans that provides a “do not eat me” signal when engrafted with human cells. Although much improved over that of the CB.17-scid strain, the NOD-scid host was still unable to support robust human T cell development following engraftment of human HSC (Shultz et al. 2007).
In the early 2000s, a major breakthrough occurred in the creation of humanized mice based on a targeted mutation in the interleukin-2-receptor γ-chain (IL2rg) locus (also known as γc and CD132) (Cao et al. 1995; DiSanto et al. 1995; Jacobs et al. 1999; Ohbo et al. 1996). When the IL2rgnull mutation was combined with the Prkdcscid or recombination activating gene 1 or 2 null mutations (Rag1null and Rag2null) in mice to create the NOD.Cg-PrkdcscidIl2rgtm1Wjl (NSG) (Shultz et al. 2005), the NODShi.Cg-PrkdcscidIl2rgtm1Sug (NOG) (Ito et al. 2002), NOD.Cg-Rag1tm1MomIL2rgtm1Wjl (NRG) (Pearson et al. 2008), and the C;129S4-Rag2tmFwa1Il2rgtm1Sug BRG strains (Traggiai et al. 2004), these mice were observed to support increased levels of human hematopoietic and immune cell engraftment, although the NSG and NRG strains supported higher levels of engraftment than the BRG strain (Brehm et al. 2010). Moreover, the NSG and NRG strains were found to support the growth of hematological cancers that could not grow in CB.17-scid or NOD-scid mice (Shultz et al. 2014). The IL2r γ-chain gene is a key molecule in the high-affinity signaling receptors for IL2, IL4, IL7, IL9, IL15, and IL21, leading to severe defects in both adaptive and innate immunity, and when combined with the Prkdcscid or the Rag1null or Rag2null mutations, create severely immunodeficient murine hosts that can be engrafted with functional human immune systems (Sugamura et al. 1996). However, when the IL2rγnull mutation was combined with immunodeficient C57BL/6 or BALB/c strain mice, much lower human cell engraftment was observed (Brehm et al. 2010; Legrand et al. 2011; Yamauchi et al. 2013). This was largely due to fact that the Sirp-α polymorphisms in these mice are very different to that of humans (Barclay and Brown 2006). This was proven by generating NOD-Sirp-α congenic C57BL/6-Rag2nullIL2rγnull (B6RGS) (Yamauchi et al. 2013), NOD Sirp-α congenic BALB/c Rag2nullIL2rγnull (BRGS) (Legrand et al. 2011), and human Sirp-α transgenic BALB/c Rag2nullIL2rγnull (BRGS) (Strowig et al. 2011) mice, which allowed high levels of human cell engraftment to be achieved. There are now multiple strains of mice with the IL2rgnull combined with either the Prkdcscid or the Rag1nullor Rag2null mutations (reviewed in (Brehm et al. 2016; Kenney et al. 2016; Theocharides et al. 2016; Walsh et al. 2017)). An overview of the published strains of immunodeficient IL2rgnull mice and their availability is shown in Table 1.
Model systems for engrafting human immune systems
There are three basic approaches to engrafting a human immune system into immunodeficient mice, all first reported in 1988 using CB.17-scid mice as hosts.
Hu-PBL-SCID
The simplest method for establishing a human immune system in immunodeficient mice is the engraftment of human peripheral blood mononuclear cells (PBMCs), termed the human peripheral blood lymphocyte SCID (Hu-PBL-SCID) model (Mosier et al. 1988). In this model, human lymphocytes engraft and rapidly expand, leading to engraftment of predominately mature CD3+ T cells with few myeloid or B cells observed by 10–14 days after engraftment (Ito et al. 2009; King et al. 2009). In addition to the homeostatic expansion of the T cells in the immunodeficient host, the majority of the T cells that engraft appear to be xeno-reactive leading to the rapid development of a lethal graft-versus-host disease (GVHD). This model is used to study human T cell activation and effector function and to evaluate immunosuppressive drugs.
Hu-SRC-SCID
The engraftment of human HSCs into CB-17-scid mice was termed the human SCID repopulating cell SCID (Hu-SRC-SCID) model (Kamel-Reid and Dick 1988), but the SCID-repopulating cell is now known to be a human CD34+ HSC. Human CD34+ HSCs can be obtained from multiple sources, including G-CSF-mobilized blood, bone marrow, umbilical cord blood, or fetal liver (Shultz et al. 2012). The efficiency of engraftment is highest when using fetal liver or cord blood CD34+ cells, but less efficient when using G-CSF mobilized blood or bone marrow CD34+ cells (reviewed in Brehm et al. 2016; Shultz et al. 2012). Hu-SRC-SCID mice develop all lineages of hematopoietic cells, including T cells, B cells, NK cells, myeloid cells, and precursors for red blood cells (RBCs), megakaryocytes and granulocytes. However, myeloid cell development is not robust, likely due to the lack of species-specific factors (Theocharides et al. 2016). Furthermore, the human RBCs, and platelets circulate poorly in humanized mice, likely due their rapid removal from the circulation by murine macrophages (Hu and Yang 2012; Suzuki et al. 2007; Willinger et al. 2011).
SCID-hu and BLT
Implantation of 16–22-week gestation fragments of human fetal liver and thymus in the subrenal capsular space is termed the SCID-hu model (McCune et al. 1988). In this model, a thymic organoid develops. In the bone marrow–liver–thymus (BLT) model, CD34+ fetal liver cells from the same donor are injected intravenously into sublethally-irradiated recipients of fetal liver and thymus fragments (Lan et al. 2006; Melkus et al. 2006). In BLT mice, the development of a human thymus organoid and the generation of a robust peripheral immune system are observed. Although the BLT model was first described in the original SCID-hu publication (McCune et al. 1988), development of a peripheral immune system was not observed, likely due to the suboptimal mouse strain (CB.17-scid) used in those experiments. When using NOD-scid mice, peripheral immune systems develop, including a human mucosal immune system (Denton et al. 2012; Kalscheuer et al. 2012) that permits mucosal routes of HIV infection to be investigated (Sun et al. 2007).
Limitations of current humanized mouse models and approaches to overcome them
While humanized mice have been used effectively to model human immunity, there remain a number of limitations. We will briefly review some of the limitations in each model system and approaches to overcome them.
Hu-PBL-SCID
The PBMC-engraftment model has been used for the investigation of GVHD (Ito et al. 2009; King et al. 2009) and other T cell-mediated immune responses such as allograft rejection (for review see Kenney et al. 2016). However, a major limitation of the model is the underlying xenogeneic GVHD that invariably leads to death (Ito et al. 2009; King et al. 2009). We have reported that the majority of this human T cell xeno-reactivity is directed against the murine MHC class I and class II molecules (King et al. 2009). To address this, investigators have developed strains of immunodeficient mice that lack both murine MHC class I and class II. NOG mice deficient in MHC class II (IAnull) were crossed to NOG-B2Mnull mice to create a NOG mouse deficient in both MHC class I and class II (Yaguchi et al. 2018). These mice displayed little GVHD when engrafted with human PBMC. Furthermore, when immunized with influenza vaccine, the mice developed antigen-specific human B cells. When challenged with HLA-A2 cytomegalovirus peptide, the development of antigen-specific human T cells was observed (Yaguchi et al. 2018). Our laboratories developed two NSG strains deficient in murine MHC class I and class II, the NOD.Cg- B2mtm1UncPrkdcscidH2dlAb1EaIl2rgtm1Wjl/Sz/SzJ, abbreviated as NSG-B2Mnull(IA IE)null and NOD.Cg-PrkdcscidH2Ab1em1MwvH2K1tmiBpeH2D1tmiBpeIl2rgtm1Wjl/Sz/SzJ, abbreviated as,NSG-(KbDb)null (IAnull) mouse strains (Brehm et al. 2018). These mice readily engraft with mature human T cells that are capable of mediating allograft rejection of human islets in the absence of the confounding effects of GVHD. The major difference between these NSG-based strains is their clearance of human IgG. The neonatal Fc receptor (FcRN) is responsible for prolonging the half-life of IgG in the blood (Roopenian and Akilesh 2007), and its expression requires B2M (Raghavan and Bjorkman 1996). In the NSG-B2Mnull(IA IE)null strain, FcRN is not expressed and human IgG half-life in the circulation is extremely short as compared to that observed in NSG-(KbDb)null (IAnull) mice, which have IgG half-life circulation that is comparable to that observed in NSG mice (Brehm et al. 2018). Thus, when testing any mAb-based therapeutics in the NOD-based MHC class I and class II knockout mice, it will be important to use the NSG-(KbDb)null (IAnull) strain. It has also recently been reported that when PBMC are engrafted into NOG mice transgenically expressing human IL4, that GVHD symptoms are significantly depressed. Moreover, immunization of these mice with the HER2 multiple antigen peptide (CH401MAP) or keyhole limpet hemocyanin (KLH) led to induction of antigen-specific IgG antibody production (Kametani et al. 2017). These new models will provide opportunities to investigate mature human T cell function in humanized mice in the absence of the confounding effects of GVHD.
Hu-SRC-SCID
Human HSC-engrafted immunodeficient IL2rγnull mice can develop a complete hematopoietic system, including human T cells, B cells, and innate immune cells, although the B cells appear to be immature and it is difficult to generate class switching (Brehm et al. 2016; Shultz et al. 2012). Moreover, the human T cells are educated on the murine MHC and thus the majority of these T cells are H2-restricted (Watanabe et al. 2009). This H2-selection makes interactions between T cells and HLA-expressing human antigen-presenting cells (APCs) difficult. To overcome this, investigators have generated a number of transgenic HLA-expressing immunodeficient IL2rγnull mice that can be engrafted with HLA-matched HSCs to permit the development of human HLA-restricted T cell responses (Akkina et al. 2016; Brehm et al. 2016, 2013, 2014; Legrand et al. 2009; Shultz et al. 2012; Theocharides et al. 2016; Walsh et al. 2017). In addition to the generation of HLA transgenic mice, investigators have used AAV vectors to induce the expression of HLA-class I (HLA-A2) (Huang et al. 2014) in NSG mice engrafted with HLA-A2 CD34+ HSCs. In this study, HLA-restricted human CD8+ T cells were observed following immunization with malaria or HIV antigens. In another study, AAV vectors were used to express HLA class II (DR1 or DR4) in NSG mice engrafted with HLA-matched CD34+ HSCs (Sharma et al. 2016). Following infection with respiratory syncytial virus (RSV), the virus was cleared faster than in NSG mice not transduced with HLA class II AAV vectors. Furthermore, anti-RSV antibodies and RSV-specific T cell production of interferon γ were observed (Sharma et al. 2016). AAV vector delivery of HLA molecules will allow rapid development of HLA-expression in the next generation immunodeficient strains of mice that have been and are continually being created without the need for creating new transgenic mice or backcrossing to achieve transgenic expression of HLA in each strain (Table 1).
A second major limitation is the lack of cytokines that promote growth and differentiation of human HSC and immune systems in mice. Many of the cytokines required for human hematopoietic and immune cell development cannot be replaced by murine cytokines due to species-specificity (Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016). To address this, human cytokines have been delivered into humanized mice by transgenic expression of cDNA constructs and BACs, knock-in technology, viral expression vectors and by plasmid delivery using hydrodynamic injections. Each of these approaches has their own unique limitations (for reviews, see Akkina et al. 2016; Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016).
Of particular interest are innate immune cytokines that are species-specific leading to suboptimal human myeloid and NK cell development in all of the standard humanized mouse models (Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016). To address this, a number of human cytokine-expressing mice have been developed (Table 1). Many of these cytokine expressing models of humanized mice have been described in reviews (Akkina et al. 2016; Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016). We will focus on the more recent cytokine expressing humanized mouse models created to enhance the development of human immune cells.
An important advancement for humanized mouse models is the generation of immunodeficient mice that have enhanced development of human innate immune cells such as myeloid cells and macrophages. Although human myeloid cells develop in human HSC-engrafted immunodeficient IL2rγnull mice, myeloid development is suboptimal and few mature human macrophages are generated (Brehm et al. 2016; Rongvaux et al. 2013; Shultz et al. 2012). A NOG model transgenically expressing human GM-CSF and IL3 has been reported to have increased levels of human myeloid cells when engrafted with human HSCs (Ito et al. 2013). A recent model has been developed that transgenically expresses human M-CSF, IL3, Sirp-alpha, and TPO on a (129xBALB/c) strain with mutated Rag2null and IL2rγnull genes and is termed MISTRG (Rongvaux et al. 2014). These mice develop robust populations of human myeloid cells. Another model is a NSG strain that transgenically expresses SCF, GM-CSF, and IL3, the NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(CMV-IL3,CSF2,KIL)1Eav/MloySzJ strain, abbreviated as (NSG-SGM3), which demonstrates increased levels of human myeloid cells when engrafted with human HSCs. These mice also generate increased numbers of regulatory T cells (Billerbeck et al. 2011; Covassin et al. 2013; Jangalwe et al. 2016). However, few mature human macrophages develop in NSG-SGM3 mice, which has been overcome in NSG-Tg(huCSF1) strain (Lee et al. 2013).
We have combined the NSG-SGM3 with the NSG-Tg(huCSF1) strain to generate NSG-SGM3 CSF1 (QUAD) mice. Engraftment of QUAD mice with human CD34+ HSCs results in increased human CD33+ myeloid development (Fig. 1a), higher levels of mature CD14+ macrophages (Fig. 1b) and increased human inflammatory cytokine release 6 h following injection with 15 µg of LPS (Fig. 1c) as compared with NSG and NSG-SGM3 human HSC-engrafted mice.

Human CD33+ (a) and CD14+ (b) myeloid cells in NSG strains of mice 12 weeks after irradiation (200 cGy) and engraftment with 105 human CD34+ HSCs as determine by flow cytometry. Plasma concentrations of human cytokines 6 h after injection of 15 µg of LPS as determined by a BD cytometric bead array (c). Each symbol represents an individual mouse. N = 3 experiments
In HSC-engrafted immunodeficient IL2rγnull mice, both plasmacytoid (CD123+) and myeloid (CD11c+) dendritic cells (DCs) are detected (Akkina et al. 2016; Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016). Based on the observation that administration of recombinant Flt3-ligand increased the CD141+ and myeloid CD1c+ DCs as well as increased the level of plasmacytoid DCs (Ding et al. 2014), investigators have performed hydrodynamic injection of Flt3 and GM-CSF into NOD-scid Jak3null mice to increase these human DC populations (Iwabuchi et al. 2018). To further enhance human DC development, the receptor for Flt3-ligand, Flk2/Flt3 was knocked out in BRG mice to create space for human DCs. Engraftment of these mice with human HSCs plus administration of recombinant Flt3-ligand enhanced human DC development (Li et al. 2016).
Another innate immune cell population that displays suboptimal development in HSC-engrafted immuodeficient mice is the human NK cell. It has been shown that injection of recombinant IL15/IL15-receptor complexes enhances the development of human NK cells in HSC-engrafted BRG mice (Huntington et al. 2009). Based on these observations, human IL15 transgenic mice have been developed. A NOG-Tg(huIL15) mouse has been reported with the human IL15 cDNA expressed under the control of the CMV promoter (Katano et al. 2017). The level of circulating IL15 in these mice is ~ 50 pg/ml and these mice support long term survival of peripheral blood -erived human NK cells that are able to impede the growth of the NK-cell sensitive K562 leukemia cell line. Another model is based on a human IL15 knock-in into a (129 × BALB/c)F1 human Sirp-α transgenic mouse (Herndler-Brandstetter et al. 2017). These mice have very low circulating levels of IL15 but following stimulation with poly I:C, they express high levels (~ 200–400 pg/ml). These mice develop robust levels of functional human NK cells that can mediate antibody-dependent cellular cytotoxicity against the human NK-resistant Raji cell line (Herndler-Brandstetter et al. 2017).
Additional strains have been created that transgenically express human IL6. Using knock-in technology, transgenic IL6 mice were constructed in (129 × BALB/c) F1 embryonic stem cells (ESCs), and the mice were intercrossed to create a (129 × BALB/c)F1-Rag2nullIL2rγnull (BRG) strain that expresses the human Sirp-α and is homozygous for the IL6 transgene (termed RG SKI IL6) (Yu et al. 2017). When engrafted with human HSCs, immunization with ovalbumin (OVA) generated human IgG anti-OVA antibodies. In another model, a NOG-Tg(huIL6)-transgenic mouse was created using human IL6 cDNA under the control of the cytomegalovirus (CMV) promoter (Hanazawa et al. 2018). When engrafted with human CD34+ cells, significant levels of HLA-DR-negative human monocytes were observed in the NOG-Tg(huIL6) transgenic mice, but not in NOG mice. Engraftment with a human head and neck squamous cell carcinoma-derived cell line resulted in intra-tumor infiltrating human CD163+ macrophages that secreted arginase-1, IL10, and VEGF, suggesting they were immunosuppressive tumor-associated macrophages (TAMs) (Hanazawa et al. 2018). Finally, an additional IL6 transgenic strain was created in the laboratory of Dr. Shultz on the NSG background using a human IL6 BAC. These mice readily engrafted with human umbilical cord blood-derived CD34+ HSCs, with levels similar to that observed in the RG SKI IL6 mice (Yu et al. 2017). Higher levels of CD3+ T cells in the blood as compared to HSC-engrafted NSG non-IL6 transgenic mice were observed (Fig. 2). IL6 is known to be a driver of IL17 cell differentiation (Fig. 3, Gaffen et al. 2014). To determine whether IL17 cells were preferentially generated in HSC-engrafted NSG-Tg(huIL6) mice, CD4+ spleen cells were stimulated with PMA and ionomycin. NSG-Tg(huIL6) mice generated higher levels of Th17 cells than did NSG CD4+ spleen cells (Fig. 4, p = 0.037). HSC-engrafted NSG-Tg(huIL6) mice also had higher plasma levels of IgM (9.1 ± 1.7 × 104 ng/ml, N = 11) as compared to NSG mice (3.0 ± 0.8 × 104 ng/ml, N = 10, p = 0.007) as well as higher levels of IgG (2.0 ± 0.2 × 103 ng/ml, N = 11, and 0.6 ± 0.2 × 103 ng/ml, N = 10, respectively, p = 0.001), 15 weeks after HSC engraftment. When infected with Dengue virus, Dengue-specific IgG was observed in NSG-Tg(huIL6) mice (N = 2) but not in Dengue virus-infected NSG mice (N = 3) (data not shown). Combined, these data suggest that HSC-engrafted human IL6-expressing immunodeficient mice will be useful models for the elicitation of antigen-specific antibody responses, investigation of the development of human Th17 cells, and the immunosuppressive mechanisms associated with TAMs.

Development of human CD45+ cells (a) and CD3+ T cells (b) in the blood of HSC-engrafted NSG and NSG-Tg(huIL6) mice. Six-week-old mice were irradiated with 200 cGy and engrafted with 50,000 human CD34+ cord blood-derived CD34+ HSCs. At various times, the levels of human cells in the blood were determined by flow cytometry. Each symbol represents an individual mouse. N = 2 experiments

Cytokine involvement in the differentiation of various CD4+ T cell subsets

Development of Th17 T cells in NSG (filled circles) and NSG-Tg(huIL6) (open squares) mice
Finally, in most HSC-engraftment models, pre-conditioning of the host, usually using low doses of irradiation, is required for engraftment of human HSCs and development of a human immune system (Akkina et al. 2016; Brehm et al. 2016; Shultz et al. 2012; Theocharides et al. 2016). However, it has been reported that NSG mice transgenically expressing membrane-bound human stem cell factor (SCF) can be engrafted with human HSCs in the absence of irradiation preconditioning (Brehm et al. 2012). More recently, a NSG model based on a W41 mutation resulting in a reduction of function in the murine Kit gene has been reported (Rahmig et al. 2016). The Kit gene regulates HSC survival, and homozygosity for the KitW41 mutation leads to a deficiency in murine HSCs, creating space for the engraftment of human HSCs in the absence of any pre-conditioning (Broudy 1997). These mice develop a human hematopoietic and immune system similar to that observed in NSG mice preconditioned with irradiation and engrafted with human HSC. Interestingly, it has been reported that HSC engraftment of the NSG-KitW41/W41 strain leads to better RBC and platelet development in the bone marrow than is observed in irradiated NSG mice (Rahmig et al. 2016).
BLT
Human BLT mice on the NSG background develop robust human immune systems, including robust mucosal immune systems (Denton et al. 2012; Kalscheuer et al. 2012). Because human T cell development occurs in the autologous HLA-expressing thymus, HLA-restricted T cells can be elicited by infection or immunization (Melkus et al. 2006). However, these mice are prone to development of a wasting GVHD-like disease in most (Covassin et al. 2013; Greenblatt et al. 2012; Lockridge et al. 2013) but not all (Onoe et al. 2011) laboratories. A new model of humanized BLT mice has been developed based on disruption of the Sirp-α/CD47 axis (Lavender et al. 2014, 2013). Genetic crosses between C57BL/6-CD47KO mice and C57BL/6-Rag2nullIL2rγnull mice resulted in Triple KO (TKO) mice. TKO-BLT mice develop robust peripheral immune systems. Moreover, it has been reported that these mice do not develop the wasting GVHD-like disease observed in NSG mice (Lavender et al. 2013, 2014). However, availability of human fetal tissues is limited, the generation of BLT mice requires technical surgical skills, and there is the potential of constraints on the use of human fetal tissue in research in the US (Editorial 2017; Wadman 2015). To begin to address this, a new “BLT-like” model has been described wherein neonatal human thymus, removed prior to cardiac surgery, and cord blood-derived CD34+ HSCs are used to set up a model similar to that of the BLT (Brown et al. 2018). This model would address the potential ethical constraints associated with using fetal tissues in research. Fragments of the recovered thymi were frozen for use. Co-engraftment of NSG mice with these thymus fragments and either autologous cord blood-derived CD34+ HSCs or allogeneic CD34+ HSCs, in combination with injection of anti-human CD2 mAb to deplete passenger thymocytes led to the generation of a peripheral human immune system similar to that observed in the BLT model. These mice were able to mount immune responses such as rejection of a C57BL/6 skin graft and a delayed hypersensitivity response (Brown et al. 2018). However, the thymic organoid was extremely small as compared to a fetal thymus organoid. The engraftment of a mucosal immune system, which is present in BLT mice, was not investigated (Brown et al. 2018). This model is still in the early stages of development and validation, and it is not known whether it will be a viable alternative to the BLT model or another humanized mouse model with its own associated strengths and weaknesses.
Lymph node development
A common limitation in all of the humanized immunodeficient IL2rγnull mouse models is the poor lymphatic architecture and suboptimal lymph node development and organization that are observed (Kenney et al. 2016; Rongvaux et al. 2013; Shultz et al. 2012; Theocharides et al. 2016).
The factors and steps important in the development of lymphoid structures and organization are summarized in Fig. 5. CXCL13 secreted by mesenchymal cells in the lymph node anlage stroma interacts with its receptor, CXCR5, on lymphoid tissue-inducer (LTi) cells (Marchesi et al. 2009). This signaling leads to LTi cell induction of mesenchymal cells in the lymph node anlage to develop a formed cell cluster that differentiates into lymphoid tissue organizer (LTo) cells, which are important in attracting and retaining hematopoietic cells in the lymph node (Weih and Caamano 2003). The importance of CXCL13 is based on the report that mice that lack CXCL13 signaling to LTi cells demonstrate loss of all peripheral lymph nodes except for mesenteric, facial, and cervical lymph nodes (Bar-Ephraim and Mebius 2016), similar to what is observed in NSG mice (Ito et al. 2002; Manz 2007; Rongvaux et al. 2014; Shultz et al. 2012, 2005; Traggiai et al. 2004). This also results in “empty” peripheral lymph nodes due to their failure to attract and become engrafted with immune cells (Carragher et al. 2004; van de Pavert et al. 2009; Weih and Caamano 2003). CXCL13 also induces CD4 cell development into follicular helper T cells (Thf) cells during antigen stimulation (Crotty 2014; Ueno et al. 2015). Thf cells provide stimulatory signals to B cells to mediate positive selection of high-affinity B cells and the differentiation of plasma cells in the germinal centers (Crotty 2014; Sage and Sharpe 2016). CXCL13 stimulation of follicular DCs is also important in organization of lymphoid structure in the spleen (Neely and Flajnik 2015, 2016). Many of these events are dependent on the IL2rγ-chain which is absent in IL2rγnull mice.

Lymphoid structure development. Lymph node anlage mesenchymal cells (MC) secrete CXCL13 that activates lymphoid tissue-inducer (LTi) cells, which in turn induces the development of lymphoid tissue organizer (LTo) cells and creation of lymphoid structures. CXCL13 also recruits T helper follicular (Thf) CD4 cells and follicular DCs to the lymphoid structure that are important in the organization of lymphoid organs. Many of the signaling pathways important in this cascade are dependent on the IL2rγ-chain which is absent in IL2rγnull mice
A number of laboratories have begun to address poor lymph node development in HSC-engrafted IL2rγnull mice with limited success to date. One of the essential cells for lymph node development is the LTi cell whose development and function is in part dependent on signaling through the high-affinity IL7 receptor (Bar-Ephraim and Mebius 2016). The IL2rγ chain is an essential part of the IL7 receptor, and in IL2rγnull mice, LTi cell development is impaired and lymph node development is suboptimal. To address this, one laboratory developed a transgenic NOG mouse in which the entire BAC mouse Rorg locus was used with the first exon of the Rorg replaced with the murine Il2rg gene. This allowed reconstitution of the γc molecule in LTi cells, but not in the rest of the hematopoietic cells. These mice displayed lymph node development and when engrafted with human CD34+ HSCs, and demonstrated an increase in T cell engraftment and serum Ig levels. To test for antigen-specific responses, the mice were crossed with the NOG-GM-CSF/IL3 stock to generate a NOG-RORγt-γc GM3 strain. Immunization with OVA led to increased antigen-specific anti-OVA IgG antibody. However, the production of IgG antibody, although detectable, was still relatively low and while T and B cell lymph node organization was improved, it was still relatively underdeveloped (Takahashi et al. 2018).
Another molecule important in LTi development and function is thymic stromal lymphopoietin (TSLP). TSLP signals through an IL7-like receptor that is created by the alpha chain of the IL7 receptor and a γc-like chain termed TSLPR such that signaling by TSLP is IL2rγ-chain (i.e., IL7) independent. The role of TSLP in lymph node development was highlighted using IL7 and IL2rγc knockout mice, which have severe deficiencies in lymph node development (Adachi et al. 1998; Cao et al. 1995). Transgenic expression of TSLP restored lymph node development, and this effect was dependent on increased numbers of LTi cells (Chappaz and Finke 2010). Based on this data, the murine Tslp gene under the control of a keratin 14 promoter was transgenically expressed in BRG mice (Li et al. 2018). These mice demonstrated restoration of murine LTi cells with improved lymph node development. Engraftment of human CD34+ HSCs led to lymph node engraftment with B cell-like areas within follicle-like structures and diffuse T cell distribution. Following immunization with keyhole limpet hemocyanin (KLH) antigen-specific IgG and T cell responses could be detected. However, these responses still remained relatively low (Li et al. 2018). These data suggest that restoration of LTi function either in the NOG-RORγt-γc GM3 strain or the BRGS strain transgenically expressing TSLP are beneficial but not sufficient for robust lymph node development and organization.
Potential uses of humanized mice in precision medicine
From the perspective of using humanized mice to investigate mechanisms underlying human immune diseases, humanized mice can be important preclinical models prior to clinical studies in humans and be a useful tool in designing precision medicine approaches. We will highlight some areas in which humanized mice are being used to study and predict human immune responses.
With the recent Nobel Prize in Physiology or Medicine in 2018 awarded to James Allison and Tasuku Honjo “for their discovery of cancer therapy by inhibition of negative immune regulation,” cancer immunotherapy has come of age. Clinically, the major breakthrough in the treatment of human tumors came with the development of the checkpoint blockade mAb ipilimumab (Yervoy) for the treatment of melanoma (Lipson and Drake 2011), followed by tremendous advances in the use of PD-1/PD-L1 blockers such as pembrolizumab for non-small cell lung cancer (NSCLC) and other cancers. There are currently more than 3000 active clinical trials testing immunotherapy treatments and > 1100 drug combination studies involving PD-1 or PD-L1 (Kaiser 2018). Disappointingly, only ~ 20–30% of patients overall respond (Kaiser 2018). The mechanisms underlying whether the patient responds or is non-responsive to checkpoint blockade therapy will be extremely difficult to identify in the clinical trials. To study potential mechanisms, many investigators have used immunodeficient mice engrafted with human tumors and human immune systems (Shultz et al. 2014).
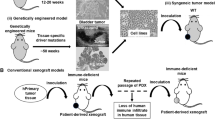
Will humanized mice recapitulate the variable response of humans to checkpoint blockade therapy observed in the clinic? We have shown the effectiveness of pembrolizumab in adult NSCLC, sarcoma, and triple-negative breast cancer (TNBC) patient-derived xenograft (PDX) tumors in HSC-engrafted NSG mice (Wang et al. 2018). In both the TNBC and the NSCLC models, pembrolizumab significantly delayed tumor growth. Of interest was the HSC donor variability in response to pembrolizumab. In both bladder cancer and NSCLC-engrafted mice, we observed in one HSC donor that pembrolizumab inhibited growth while a second HSC donor did not lead to growth inhibition of the same tumor (Wang et al. 2018). These data suggest that the PDX-humanized mouse model may reflect the variable response of adult patients to treatment with checkpoint inhibitors and may represent an approach to identify the underlying basis for this variability that could be translated to the clinic.
PDX-engrafted immunodeficient mice have been used for precision medicine in clinical trials (Hidalgo et al. 2011). In one study, the response of tumors from 14 patients were tested in immunodeficient mice with 232 different drug combinations. An effective treatment regimen was identified for 12 patients. One died before receiving treatment and durable responses were observed in 11 patients. No effective treatment was found for two patients. This work formed the basis of a company whose efforts were summarized in an article in the journal Science in 2014 (Couzin-Frankel 2014). However, it remains to be determined whether translation of this approach directly into the clinic will be predictive of clinical outcome. NCI has recently indicated a particular interest in this question and in a recent RFA, PAR-17-244 announced in November 16, 2018, a stated goal was: “develop and test novel “humanizing” approaches for mammalian models as recipients of human transplants from tumors, metastatic deposits, or early lesions”. One goal of this PAR is to solicit projects proposing to determine whether immunodeficient mice engrafted with human immune systems and human tumors will be a predictive preclinical model, particularly with respect to immunotherapies, for accelerating translation of new drugs to the clinic.
One of the major problems translating new mAb-based therapies into the clinic is the potential for development of a cytokine release syndrome (CRS). Following the severe reaction of volunteers to a mAb-based drug called TGN1412 (TeGenero) in 2006 (Suntharalingam et al. 2006), there has been a major effort to develop an animal model that would be predictive for CRS. In this study, even testing the drug in non-human primates (NHP) failed to reveal its potential to induce CRS in humans as the target molecule is not expressed on the NHP T cell population that is responsible for the human CRS response to TGN1412 (Weissmuller et al. 2016). In vitro cytokine release assays have been developed, but they may fail to accurately predict CRS in the clinic (Thorpe et al. 2013). A number of laboratories have attempted to develop a humanized mouse model of CRS based on injection of PBMC. One of the first attempts used the CB17-scid mouse that did generate a low inflammatory response following injection of OKT3 (Malcolm et al. 2012). Another laboratory used PBMC-engrafted NSG mice, and OKT3 injection led to relatively robust levels of IFNγ (Brady et al. 2014). Finally, PBMC-engrafted NRG mice lacking murine MHC class II and injected with OKT3 gave high levels of IFNγ and IL2 but only low levels of other inflammatory cytokines (Weissmuller et al. 2016). Although these models have provided “proof of principle” that humanized mice could ultimately be used to model CRS, a robust humanized mouse model for CRS remains to be developed.
Additional humanized mouse models for studies of drug therapy for anaphylactic reactions (Bryce et al. 2016; Burton et al. 2017) have been described that can be used as models for testing the efficacy of drugs to prevent anaphylactic reactions. Similarly, a number of laboratories are attempting to develop humanized mouse models of type 1 diabetes (T1D). These include engraftment of autoreactive T cell clones from individuals with T1D into HLA-matched recipients (Unger et al. 2012) as well as autoreactive TCR transduced CD4 (Ali et al. 2016) or CD8 (Babad et al. 2015) T cells injected into NSG-HLA-matched transgenic mice. In addition, human autoantigen-pulsed CD4 cells from T1D donors injected into streptozotocin-treated NSG-HLA-DR4 transgenic recipients developed insulitis but not overt diabetes (Viehmann Milam et al. 2014). In a complex BLT-engraftment model of streptozotocin-treated NSG-HLA-DQ8 mice that are also recipients of autologous BLT-derived CD8 T cells transduced with an anti-insulin TCR and immunized with insulin, 4 of 7 mice developed hyperglycemia, suggesting the development of an anti-insulin T cell response capable of killing the beta cells (Tan et al. 2017). However, a simple, reproducible precision medicine humanized mouse model of T1D remains to be developed.
Humanized mice will be important in the field of regenerative medicine as human ESC and induced pluripotent stem cells (iPSC) are considered for cell and tissue replacement in the clinic. Immunodeficient mice will be the “go to” animal model for testing the safety and efficacy of human cells and tissues derived from ESC and iPSC prior to advancing to the clinic. This could represent true “precision medicine” as each cell treatment theoretically could be an individual’s own cells and tissues derived from their iPSCs. There have been great strides in making this a reality, particularly in the diabetes field. Numerous laboratories have reported on the ability to make beta-like cells from ESCs and iPSCs and have used immunodeficient mice to test their in vivo functionality (Hosoya 2012; Maehr et al. 2009; Nair and Hebrok 2015; Nostro et al. 2015; Pagliuca et al. 2014; Rezania et al. 2013; Shahjalal et al. 2014; Zhang et al. 2009). However, to test their function in the presence of an allogeneic or autologous T1D autoimmune immune system, humanized mice are currently the only preclinical animal model available as any other immunocompetent animal model would effectively be testing a potent xenogeneic response rather than an allogeneic or autoimmune response to the human cells.
iPSCs will also be important in developing animal models of human disease. Again, taking T1D as an example, the NIH Human Islet Research Network (HIRN) consortium posted an RFA on December 17, 2018, to create animal models of T1D in the Consortium on Modeling Autoimmune Interactions (HIRN-CMAI, RFA-18-013). A number of groups that are part of the current HIRN-CMAI consortia are developing humanized mouse models of T1D based on the use of iPSCs derived from T1D individuals. For this model, T1D iPSCs are differentiated into the three key cell components underlying the cellular basis for human T1D; the target beta cell, HSCs that will generate an autoreactive immune system, and thymic epithelial cells (TECs) that will educate the autoreactive T cells that mediate the final destruction of the beta cells (Fig. 6). The T1D iPSC-derived HSCs will also generate additional immune cells such as NK cells, macrophages, and antigen-presenting cells thought to be important in the disease process. Although “proof of principle” experiments have succeeded in generating human ESC-derived HSCs that can engraft into immunodeficient mice (Sugimura et al. 2017), additional advances are needed to allow these stem cell-derived HSCs (SC-HSC) to generate robust human immune systems in the recipients. While waiting for robust immune system development in immunodeficient mice using SC-HSCs, T1D patient bone marrow has been used as a source of the autoimmune-prone HSC (Kalscheuer et al. 2012). These three cell components, SC-beta cells, SC-HSCs, and SC-TECs could then be engrafted into a single immunodeficient mouse to permit investigation of the disease process in vivo and provide a mechanistic understanding of the causes and progression of T1D (Fig. 6). These insights would make it possible to design therapeutic approaches that target cells, genes, or antigens that can induce tolerance, prevent recurrence, and possibly prevent the disease outright. Since an iPSC-based model system could be established using iPSCs derived from individuals with any immune-mediated disease, such an approach could lead to a true patient-specific precision medicine for a variety of human diseases.

Modeling T1D using SC-beta cells, SC-HSCs, and SC-TECs derived from T1D donors engrafted into immunodeficient mice
Conclusions
As our understanding of the genetic basis for human diseases continues to increase, humanized mice are poised to have a critical role for the evaluation of the biology associated with each disease, and for designing specific therapies and evaluating their efficacy prior to translation into the clinic. It must be cautioned, however, that humanized mice are models of human disease and as with all models, they have strengths as well as weaknesses. None of the currently available humanized mouse models completely recapitulate the human immune system (Brehm et al. 2016; Shultz et al. 2012). However, with an understanding of their strengths and weaknesses, humanized mice can be used as pre-clinical models to address specific questions in human biology and to guide precision medicine approaches in the clinic.
References
Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S (1998) Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150
Akkina R, Allam A, Balazs AB, Blankson JN, Burnett JC, Casares S, Garcia JV, Hasenkrug KJ, Kashanchi F, Kitchen SG, Klein F, Kumar P, Luster AD, Poluektova LY, Rao M, Sanders-Beer BE, Shultz LD, Zack JA (2016) Improvements and limitations of humanized mouse models for HIV research: NIH/NIAID “meet the experts” 2015 workshop summary. AIDS Res Hum Retroviruses 32:109–119
Ali R, Babad J, Follenzi A, Gebe JA, Brehm MA, Nepom GT, Shultz LD, Greiner DL, DiLorenzo TP (2016) Genetically modified human CD4(+) T cells can be evaluated in vivo without lethal graft-versus-host disease. Immunology 148:339–351
Babad J, Mukherjee G, Follenzi A, Ali R, Roep BO, Shultz LD, Santamaria P, Yang OO, Goldstein H, Greiner DL, DiLorenzo TP (2015) Generation of beta cell-specific human cytotoxic T cells by lentiviral transduction and their survival in immunodeficient human leucocyte antigen-transgenic mice. Clin Exp Immunol 179:398–413
Barclay AN, Brown MH (2006) The SIRP family of receptors and immune regulation. Nat Rev Immunol 6:457–464
Bar-Ephraim YE, Mebius RE (2016) Innate lymphoid cells in secondary lymphoid organs. Immunol Rev 271:185–199
Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A (2011) Development of human CD4+ FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood 117:3076–3086
Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA (1996) Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA 93:10285–10290
Bosma GC, Custer RP, Bosma MJ (1983) A severe combined immunodeficiency mutation in the mouse. Nature 301:527–530
Brady JL, Harrison LC, Goodman DJ, Cowan PJ, Hawthorne WJ, O’Connell PJ, Sutherland RM, Lew AM (2014) Preclinical screening for acute toxicity of therapeutic monoclonal antibodies in a hu-SCID model. Clin Transl Immunol 3:e29
Brehm MA, Cuthbert A, Yang C, Miller DM, DiIorio P, Laning J, Burzenski L, Gott B, Foreman O, Kavirayani A, Herlihy M, Rossini AA, Shultz LD, Greiner DL (2010) Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin Immunol 135:84–98
Brehm MA, Racki WJ, Leif J, Burzenski L, Hosur V, Wetmore A, Gott B, Herlihy M, Ignotz R, Dunn R, Shultz LD, Greiner DL (2012) Engraftment of human HSC in non-irradiated newborn NOD-scid IL2rgammanull mice is enhanced by transgenic expression of membrane-bound human SCF. Blood 119:2778–2788
Brehm MA, Jouvet N, Greiner DL, Shultz LD (2013) Humanized mice for the study of infectious diseases. Curr Opin Immunol 25:428–435
Brehm MA, Wiles MV, Greiner DL, Shultz LD (2014) Generation of improved humanized mouse models for human infectious diseases. J Immunol Methods 410:3–17
Brehm MA, Bortell R, Verma M, Shultz LD, Greiner DL (2016) Humanized mice in translational immunology. In: Tan SL (ed) Translational immunology: mechanisms and pharmacological approaches. Elsevier, Amsterdam, pp 285–326
Brehm MA, Kenney LL, Wiles MV, Low BE, Tisch RM, Burzenski L, Mueller C, Greiner DL, Shultz LD (2018) Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J e-pub ahead of print:fj201800636R
Broudy VC (1997) Stem cell factor and hematopoiesis. Blood 90:1345–1364
Brown EJ, Frazier WA (2001) Integrin-associated protein (CD47) and its ligands. Trends Cell Biol 11:130–135
Brown ME, Zhou Y, McIntosh BE, Norman IG, Lou HE, Biermann M, Sullivan JA, Kamp TJ, Thomson JA, Anagnostopoulos PV, Burlingham WJ (2018) A humanized mouse model generated using surplus neonatal tissue. Stem Cell Rep 10:1175–1183
Bryce PJ, Falahati R, Kenney LL, Leung J, Bebbington C, Tomasevic N, Krier RA, Hsu CL, Shultz LD, Greiner DL, Brehm MA (2016) Humanized mouse model of mast cell-mediated passive cutaneous anaphylaxis and passive systemic anaphylaxis. J Allergy Clin Immunol 138:769–779
Burton OT, Stranks AJ, Tamayo JM, Koleoglou KJ, Schwartz LB, Oettgen HC (2017) A humanized mouse model of anaphylactic peanut allergy. J Allergy Clin Immunol 139:314–322 e319
Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET et al (1995) Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 2:223–238
Carragher D, Johal R, Button A, White A, Eliopoulos A, Jenkinson E, Anderson G, Caamano J (2004) A stroma-derived defect in NF-kappaB2−/− mice causes impaired lymph node development and lymphocyte recruitment. J Immunol 173:2271–2279
Chappaz S, Finke D (2010) The IL-7 signaling pathway regulates lymph node development independent of peripheral lymphocytes. J Immunol 184:3562–3569
Choo JK, Seebach JD, Nickeleit V, Shimizu A, Lei H, Sachs DH, Madsen JC (1997) Species differences in the expression of major histocompatibility complex class II antigens on coronary artery endothelium: implications for cell-mediated xenoreactivity. Transplantation 64:1315–1322
Couzin-Frankel J (2014) Hope in a mouse. Science 346:28–29
Covassin L, Jangalwe S, Jouvet N, Laning J, Burzenski L, Shultz LD, Brehm MA (2013) Human immune system development and survival of non-obese diabetic (NOD)-scid IL2rgamma(null) (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin Exp Immunol 174:372–388
Crotty S (2014) T follicular helper cell differentiation, function, and roles in disease. Immunity 41:529–542
Davis MM (2012) Immunology taught by humans. Sci Transl Med 4:117fs112
DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2:e1600200
Denton PW, Nochi T, Lim A, Krisko JF, Martinez-Torres F, Choudhary SK, Wahl A, Olesen R, Zou W, Di Santo JP, Margolis DM, Garcia JV (2012) IL-2 receptor gamma-chain molecule is critical for intestinal T-cell reconstitution in humanized mice. Mucosal Immunol 5:555–566
Ding Y, Wilkinson A, Idris A, Fancke B, O’Keeffe M, Khalil D, Ju X, Lahoud MH, Caminschi I, Shortman K, Rodwell R, Vuckovic S, Radford KJ (2014) FLT3-ligand treatment of humanized mice results in the generation of large numbers of CD141+ and CD1c+ dendritic cells in vivo. J Immunol 192:1982–1989
DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K (1995) Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci USA 92:377–381
Donini C, D’Ambrosio L, Grignani G, Aglietta M, Sangiolo D (2018) Next generation immune-checkpoints for cancer therapy. J Thorac Dis 10:S1581–S1601
Editorial (2017) Fantasy politics over fetal-tissue research. Nature 541:133
Ellis LM, Reardon DA (2009) Cancer: the nuances of therapy. Nature 458:290–292
Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14:585–600
Greenblatt MB, Vbranac V, Tivey T, Tsang K, Tager AM, Aliprantis AO (2012) Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE 7:e44664
Greiner DL, Shultz LD (1998) Use of NOD/LtSz-scid/scid mice in biomedical research. In: Leiter EH, Atkinson MA (eds) NOD mice and related strains: research applications in diabetes, AIDS, cancer and other diseases. R.G. Landes Co., Austin, pp 173–204
Hagai T, Chen X, Miragaia RJ, Rostom R, Gomes T, Kunowska N, Henriksson J, Park JE, Proserpio V, Donati G, Bossini-Castillo L, Vieira Braga FA, Naamati G, Fletcher J, Stephenson E, Vegh P, Trynka G, Kondova I, Dennis M, Haniffa M, Nourmohammad A, Lassig M, Teichmann SA (2018) Gene expression variability across cells and species shapes innate immunity. Nature 563:197–202
Hanazawa A, Ito R, Katano I, Kawai K, Goto M, Suemizu H, Kawakami Y, Ito M, Takahashi T (2018) Generation of human immunosuppressive myeloid cell populations in human interleukin-6 transgenic NOG mice. Front Immunol 9:152
Herndler-Brandstetter D, Shan L, Yao Y, Stecher C, Plajer V, Lietzenmayer M, Strowig T, de Zoete MR, Palm NW, Chen J, Blish CA, Frleta D, Gurer C, Macdonald LE, Murphy AJ, Yancopoulos GD, Montgomery RR, Flavell RA (2017) Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc Natl Acad Sci USA 114:E9626–E9634
Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De OE, Rubio-Viqueira B, Strawn S, Wick MJ, Martell J, Sidransky D (2011) A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther 10:1311–1316
Hosoya M (2012) Preparation of pancreatic beta-cells from human iPS cells with small molecules. Islets 4:249–252
Hu Z, Yang YG (2012) Full reconstitution of human platelets in humanized mice after macrophage depletion. Blood 120:1713–1716
Huang J, Li X, Coelho-dos-Reis JG, Wilson JM, Tsuji M (2014) An AAV vector-mediated gene delivery approach facilitates reconstitution of functional human CD8+ T cells in mice. PLoS ONE 9:e88205
Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, Corcuff E, Mortier E, Jacques Y, Spits H, Di Santo JP (2009) IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med 206:25–34
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T (2002) NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100:3175–3182
Ito R, Katano I, Kawai K, Hirata H, Ogura T, Kamisako T, Eto T, Ito M (2009) Highly sensitive model for xenogenic GVHD using severe immunodeficient NOG mice. Transplantation 87:1654–1658
Ito R, Takahashi T, Katano I, Kawai K, Kamisako T, Ogura T, Ida-Tanaka M, Suemizu H, Nunomura S, Ra C, Mori A, Aiso S, Ito M (2013) Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J Immunol 191:2890–2899
Iwabuchi R, Ikeno S, Kobayashi-Ishihara M, Takeyama H, Ato M, Tsunetsugu-Yokota Y, Terahara K (2018) Introduction of human Flt3-L and GM-CSF into humanized mice enhances the reconstitution and maturation of myeloid dendritic cells and the development of Foxp3(+)CD4(+) T cells. Front Immunol 9:1042
Jacobs H, Krimpenfort P, Haks M, Allen J, Blom B, Demolliere C, Kruisbeek A, Spits H, Berns A (1999) PIM1 reconstitutes thymus cellularity in interleukin 7- and common gamma chain-mutant mice and permits thymocyte maturation in Rag- but not CD3gamma-deficient mice. J Exp Med 190:1059–1068
Jangalwe S, Shultz LD, Mathew A, Brehm MA (2016) Improved B cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun Inflamm Dis 4:427–440
Kaiser J (2018) Too much of a good thing? Science 359:1346–1347
Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, Greenberg E, Spitzer TR, Savage DG, Tahara H, Choi G, Yang YG, Sykes M (2012) A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med 4:125ra130
Kamel-Reid S, Dick JE (1988) Engraftment of immune-deficient mice with human hematopoietic stem cells. Science 242:1706–1709
Kametani Y, Katano I, Miyamoto A, Kikuchi Y, Ito R, Muguruma Y, Tsuda B, Habu S, Tokuda Y, Ando K, Ito M (2017) NOG-hIL-4-Tg, a new humanized mouse model for producing tumor antigen-specific IgG antibody by peptide vaccination. PLoS ONE 12:e0179239
Katano I, Nishime C, Ito R, Kamisako T, Mizusawa T, Ka Y, Ogura T, Suemizu H, Kawakami Y, Ito M, Takahashi T (2017) Long-term maintenance of peripheral blood derived human NK cells in a novel human IL-15- transgenic NOG mouse. Sci Rep 7:17230
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Kenney LL, Shultz LD, Greiner DL, Brehm MA (2016) Humanized mouse models for transplant immunology. Am J Transpl 16:389–397
King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L, Chase T, Gott B, Rossini AA, Bortell R, Shultz LD, Greiner DL (2009) Hu-PBL-NOD-scid IL2rgnull mouse model of xenogeneic graft-versus-host-like disease and the role of host MHC. Clin Exp Immunol 157:104–118
Klausen U, Jorgensen NGD, Grauslund JH, Holmstrom MO, Andersen MH (2019) Cancer immune therapy for lymphoid malignancies: recent advances. Semin Immunopathol 41:111–124
Koyanagi Y, Tanaka Y, Tanaka R, Misawa N, Kawano Y, Tanaka T, Miyasaka M, Ito M, Ueyama Y, Yamamoto N (1997) High levels of viremia in hu-PBL-NOD-scid mice with HIV-1 infection. Leukemia 11(Suppl 3):109–112
Lan P, Tonomura N, Shimizu A, Wang S, Yang YG (2006) Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 108:487–492
Landgraf M, McGovern JA, Friedl P, Hutmacher DW (2018) Rational design of mouse models for cancer research. Trends Biotechnol 36:242–251
Lavender KJ, Pang WW, Messer RJ, Duley AK, Race B, Phillips K, Scott D, Peterson KE, Chan CK, Dittmer U, Dudek T, Allen TM, Weissman IL, Hasenkrug KJ (2013) BLT-humanized C57BL/6 Rag2−/−gammac−/−CD47−/− mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood 122:4013–4020
Lavender KJ, Messer RJ, Race B, Hasenkrug KJ (2014) Production of bone marrow, liver, thymus (BLT) humanized mice on the C57BL/6 Rag2(−/−)gammac(−/−)CD47(−/−) background. J Immunol Methods 407:127–134
Lee J, Brehm MA, Greiner D, Shultz LD, Kornfeld H (2013) Engrafted human cells generate adaptive immune responses to Mycobacterium bovis BCG infection in humanized mice. BMC Immunol 14:53
Lee A, Sun S, Sandler A, Hoang T (2018) Recent progress in therapeutic antibodies for cancer immunotherapy. Curr Opin Chem Biol 44:56–65
Legrand N, Ploss A, Balling R, Becker PD, Borsotti C, Brezillon N, Debarry J, de JY, Deng, Di Santo H, Eisenbarth JP, Eynon S, Flavell E, Guzman RA, Huntington CA, Kremsdorf ND, Manns D, Manz MP, Mention MG, Ott JJ, Rathinam M, Rice C, Rongvaux CM, Stevens A, Spits S, Strick-Marchand H, Takizawa H, van Lent H, Wang AU, Weijer C, Willinger K, Ziegler T P (2009) Humanized mice for modeling human infectious disease: challenges, progress, and outlook. Cell Host Microbe 6:5–9
Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, de Geus SJ, Pouw SM, Bohne M, Voordouw A, Weijer K, Di Santo JP, Spits H (2011) Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer-(NK) cell homeostasis in vivo. Proc Natl Acad Sci USA 108:13224–13229
Li Y, Mention JJ, Court N, Masse-Ranson G, Toubert A, Spits H, Legrand N, Corcuff E, Strick-Marchand H, Santo JP (2016) A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur J Immunol 46:1291–1299
Li Y, Masse-Ranson G, Garcia Z, Bruel T, Kok A, Strick-Marchand H, Jouvion G, Serafini N, Lim AI, Dusseaux M, Hieu T, Bourgade F, Toubert A, Finke D, Schwartz O, Bousso P, Mouquet H, Di Santo JP (2018) A human immune system mouse model with robust lymph node development. Nat Methods 15:623–630
Lipson EJ, Drake CG (2011) Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res 17:6958–6962
Lockridge JL, Zhou Y, Becker YA, Ma S, Kenney SC, Hematti P, Capitini CM, Burlingham WJ, Gendron-Fitzpatrick A, Gumperz JE (2013) Mice engrafted with human fetal thymic tissue and hematopoietic stem cells develop pathology resembling chronic graft-versus-host disease. Biol Blood Marrow Transpl 19:1310–1322
Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA (2009) Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci USA 106:15768–15773
Malcolm SL, Smith EL, Bourne T, Shaw S (2012) A humanised mouse model of cytokine release: comparison of CD3-specific antibody fragments. J Immunol Methods 384:33–42
Manz MG (2007) Human-hemato-lymphoid-system mice: opportunities and challenges. Immunity 26:537–541
Marchesi F, Martin AP, Thirunarayanan N, Devany E, Mayer L, Grisotto MG, Furtado GC, Lira SA (2009) CXCL13 expression in the gut promotes accumulation of IL-22-producing lymphoid tissue-inducer cells, and formation of isolated lymphoid follicles. Mucosal Immunol 2:486–494
McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL (1988) The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science 241:1632–1639
Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, Garcia JV (2006) Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 12:1316–1322
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738
Mosier DE, Gulizia RJ, Baird SM, Wilson DB (1988) Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 335:256–259
Nair G, Hebrok M (2015) Islet formation in mice and men: lessons for the generation of functional insulin-producing beta-cells from human pluripotent stem cells. Curr Opin Genet Dev 32:171–180
Neely HR, Flajnik MF (2015) CXCL13 responsiveness but not CXCR5 expression by late transitional B cells initiates splenic white pulp formation. J Immunol 194:2616–2623
Neely HR, Flajnik MF (2016) Emergence and evolution of secondary lymphoid organs. Annu Rev Cell Dev Biol 32:693–711
Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, Greiner DL, Keller G (2015) Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep 4:591–604
Ohbo K, Suda T, Hashiyama M, Mantani A, Ikebe M, Miyakawa K, Moriyama M, Nakamura M, Katsuki M, Takahashi K, Yamamura K, Sugamura K (1996) Modulation of hematopoiesis in mice with a truncated mutant of the interleukin-2 receptor gamma chain. Blood 87:956–967
Onoe T, Kalscheuer H, Danzl N, Chittenden M, Zhao G, Yang YG, Sykes M (2011) Human natural regulatory T cell development, suppressive function, and postthymic maturation in a humanized mouse model. J Immunol 187:3895–3903
Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA (2014) Generation of functional human pancreatic beta cells in vitro. Cell 159:428–439
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47
Pearson T, Shultz LD, Miller D, King M, Laning J, Fodor W, Cuthbert A, Burzenski L, Gott B, Lyons B, Foreman O, Rossini AA, Greiner DL (2008) Non-obese diabetic-recombination activating gene-1 (NOD-Rag1 null) interleukin (IL)-2 receptor common gamma chain (IL2r gamma null) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol 154:270–284
Perrin S (2014) Preclinical research: make mouse studies work. Nature 507:423–425
Raghavan M, Bjorkman PJ (1996) Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol 12:181–220
Rahmig S, Kronstein-Wiedemann R, Fohgrub J, Kronstein N, Nevmerzhitskaya A, Bornhauser M, Gassmann M, Platz A, Ordemann R, Tonn T, Waskow C (2016) Improved human erythropoiesis and platelet formation in humanized NSGW41 Mice. Stem Cell Rep 7:591–601
Rezania A, Bruin JE, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ (2013) Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 31:2432–2442
Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, Manz MG (2013) Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol 31:635–674
Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, Manz MG, Flavell RA (2014) Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol 32:364–372
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–725
Rosenthal N, Brown S (2007) The mouse ascending: perspectives for human-disease models. Nat Cell Biol 9:993–999
Sage PT, Sharpe AH (2016) T follicular regulatory cells. Immunol Rev 271:246–259
Sato K, Takeuchi JS, Misawa N, Izumi T, Kobayashi T, Kimura Y, Iwami S, Takaori-Kondo A, Hu WS, Aihara K, Ito M, An DS, Pathak VK, Koyanagi Y (2014) APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PLoS Pathog 10:e1004453
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation, Host Response to Injury LSCRP (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 110:3507–3512
Shahjalal HM, Shiraki N, Sakano D, Kikawa K, Ogaki S, Baba H, Kume K, Kume S (2014) Generation of insulin-producing beta-like cells from human iPS cells in a defined and completely xeno-free culture system. J Mol Cell Biol 6:394–408
Sharma A, Wu W, Sung B, Huang J, Tsao T, Li X, Gomi R, Tsuji M, Worgall S (2016) Respiratory syncytial virus (RSV) pulmonary infection in humanized mice induces human anti-RSV immune responses and pathology. J Virol 90:5068–5074
Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL (1995) Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol 154:180–191
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2rgnull mice engrafted with mobilized human hematopoietic stem cell. J Immunol 174:6477–6489
Shultz LD, Ishikawa F, Greiner DL (2007) Humanized mice in translational biomedical research. Nat Rev Immunol 7:118–130
Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 12:786–798
Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL (2014) Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harb Protoc 2014:694–708
Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, Eynon EE, Manz MG, Flavell RA (2011) Transgenic expression of human signal regulatory protein alpha in Rag2-/-γc-/- mice improves engraftment of human hematopoietic cells in humanized mice. Proc Natl Acad Sci USA 108:13218–13223
Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, Nakamura M, Takeshita T (1996) The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol 14:179–205
Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, Goettel JA, Serrao E, Rowe RG, Malleshaiah M, Wong I, Sousa P, Zhu TN, Ditadi A, Keller G, Engelman AN, Snapper SB, Doulatov S, Daley GQ (2017) Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545:432–438
Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, Melkus MW, Padgett-Thomas A, Zupancic M, Haase AT, Garcia JV (2007) Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med 204:705–714
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355:1018–1028
Suzuki K, Hiramatsu H, Fukushima-Shintani M, Heike T, Nakahata T (2007) Efficient assay for evaluating human thrombopoiesis using NOD/SCID mice transplanted with cord blood CD34+ cells. Eur J Haematol 78:123–130
Takahashi T, Katano I, Ito R, Goto M, Abe H, Mizuno S, Kawai K, Sugiyama F, Ito M (2018) Enhanced antibody responses in a novel NOG transgenic mouse with restored lymph node organogenesis. Front Immunol 8:2017
Takao K, Miyakawa T (2014) Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA 112:1167–1172
Takao K, Miyakawa T (2015) Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA 112:1167–1172
Takao K, Hagihara H, Miyakawa T (2014) Reply to Warren et al. and Shay et al.: Commonalities across species do exist and are potentially important. Proc Natl Acad Sci USA 112:E347–E348
Takao K, Hagihara H, Miyakawa T (2015) Reply to Warren et al. and Shay et al.: Commonalities across species do exist and are potentially important. Proc Natl Acad Sci USA 112:E347–E348
Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, Li Y, Khosravi Maharlooei M, Chavez E, Nauman G, Danzl N, Nakayama M, Roep BO, Sykes M, Yang YG (2017) Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci USA 114:10954–10959
Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG (2016) Humanized hemato-lymphoid system mice. Haematologica 101:5–19
Thorpe SJ, Stebbings R, Findlay L, Eastwood D, Poole S, Thorpe R (2013) How predictive are in vitro assays for cytokine release syndrome in vivo? A comparison of methods reveals worrying differences in sensitivity and frequency of response. Cytokine 64:471–472
Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG (2004) Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 304:104–107
Ueno H, Banchereau J, Vinuesa CG (2015) Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol 16:142–152
Unger WW, Pearson T, Abreu JR, Laban S, van der Slik AR, der Kracht SM, Kester MG, Serreze DV, Shultz LD, Griffioen M, Drijfhout JW, Greiner DL, Roep BO (2012) Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/SCID/IL2RG null mice. PLoS ONE 7:e49213
van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, Kusser K, Hopken UE, Lipp M, Niederreither K, Blomhoff R, Sitnik K, Agace WW, Randall TD, de Jonge WJ, Mebius RE (2009) Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol 10:1193–1199
Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, Herold KC, Bothwell AL (2014) A humanized mouse model of autoimmune insulitis. Diabetes 63:1712–1724
Wadman M (2015) The truth about fetal tissue research. Nature 528:178–181
Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, Shultz LD (2017) Humanized mouse models of clinical disease. Annu Rev Pathol 12:187–215
Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, Shi W, Ma AH, De Vere White RW, Airhart S, Liu ET, Banchereau J, Brehm MA, Greiner DL, Shultz LD, Palucka K, Keck JG (2018) Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J 32:1537–1549
Warren HS, Tompkins RG, Moldawer LL, Seok J, Xu W, Mindrinos MN, Maier RV, Xiao W, Davis RW (2014) Mice are not men. Proc Natl Acad Sci USA 112:E345
Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, Ito R, Ito M, Minegishi M, Minegishi N, Tsuchiya S, Sugamura K (2009) The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice). Int Immunol 21:843–858
Weih F, Caamano J (2003) Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev 195:91–105
Weissmuller S, Kronhart S, Kreuz D, Schnierle B, Kalinke U, Kirberg J, Hanschmann KM, Waibler Z (2016) TGN1412 induces lymphopenia and human cytokine release in a humanized mouse model. PLoS ONE 11:e0149093
Wilkinson RW, Leishman AJ (2018) Further advances in cancer immunotherapy: going beyond checkpoint blockade. Front Immunol 9:1082
Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, Auerbach W, Eynon EE, Stevens S, Manz MG, Flavell RA (2011) Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc Natl Acad Sci USA 108:2390–2395
Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, Iwata T, Ka Y, Katano I, Ito R, Ito M, Kawakami Y (2018) Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol 15:953–962
Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, Iwamoto C, Nishihara M, Iwasaki H, Miyamoto T, Honma N, Nakao M, Matozaki T, Akashi K (2013) Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood 121:1316–1325
Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, Frleta D, Gurer C, Murphy AJ, Yancopoulos GD, Meffre E, Manz MG, Flavell RA (2017) A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood 129:959–969
Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H (2009) Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res 19:429–438
Acknowledgements
We thank Zoe Reifsnyder for assistance in preparation of the figures. This work was supported, in part, by U.S. National Institutes of Health (NIH) Office of the Director Grant 1R24 OD018259 (M.A.B., D.L.G., and L.D.S.), and the NIH National Institute of Diabetes and Digestive and Kidney Diseases—supported Human Islet Research Network (https://hirnetwork.org) Grants and NIH UC4 DK104218 (M.A.B., D.L.G., and L.D.S.), CA034196 (to L.D.S.), 1R01 AI132963 (M.A.B. and L.D.S.), 1DP3DK111898 (M.A.B.), and 1R01 DK1035486 (M.A.B.) grants. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M.A.B. and D.L.G. are consultants for The Jackson Laboratory. The remaining authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shultz, L.D., Keck, J., Burzenski, L. et al. Humanized mouse models of immunological diseases and precision medicine. Mamm Genome 30, 123–142 (2019). https://doi.org/10.1007/s00335-019-09796-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-019-09796-2