
Abstract
To determine annotations of the sequence elements on microarrays used for transcriptional profiling experiments in livestock species, currently researchers must either use the sparse direct annotations available for these species or create their own annotations. ANEXdb (http://www.anexdb.org) is an open-source web application that supports integrated access of two databases that house microarray expression (ExpressDB) and EST annotation (AnnotDB) data. The expression database currently supports storage and querying of Affymetrix-based expression data as well as retrieval of experiments in a form ready for NCBI-GEO submission; these services are available online. AnnotDB currently houses a novel assembly of approximately 1.6 million unique porcine-expressed sequence reads called the Iowa Porcine Assembly (IPA), which consists of 140,087 consensus sequences, the Iowa Tentative Consensus (ITC) sequences, and 103,888 singletons. The IPA has been annotated via transfer of information from homologs identified through sequence alignment to NCBI RefSeq. These annotated sequences have been mapped to the Affymetrix porcine array elements, providing annotation for 22,569 of the 23,937 (94%) porcine-specific probe sets, of which 19,253 (80%) are linked to an NCBI RefSeq entry. The ITC has also been mined for sequence variation, providing evidence for up to 202,383 SNPs, 62,048 deletions, and 958 insertions in porcine-expressed sequence. These results create a single location to obtain porcine annotation of and sequence variation in differently expressed genes in expression experiments, thus permitting possible identification of causal variants in such genes of interest. The ANEXdb application is open source and available from SourceForge.net.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microarray and other high-throughput expression platforms can provide a vast amount of information about transcriptional products within a biological sample under a variety of conditions. It is essential that functions be assigned to these transcripts so that transcriptome data can be fully utilized to explore important biological questions. Many human and mouse transcripts have functional annotations (e.g., currently there are 11,616 manually annotated human proteins and 14,182 manually annotated mouse proteins with GO terms through the GOA project; see http://www.ebi.ac.uk/GOA/human_release.html and http://www.ebi.ac.uk/GOA/mouse_release.html for results on both manual and automated GO annotations, released July 2009); most of these are indirect annotations predicted from structural similarity to genes and proteins in prokaryotes and lower eukaryotes. However, for species such as the pig, relatively little direct or indirect annotation is available for most expressed sequences (ESTs). One way to address this problem is to leverage sequence annotations from the better-annotated species, e.g., mouse, to infer annotations of their homologs in other species, e.g., pig (Nagaraj et al. 2006).
Such an approach to porcine EST annotation requires that as many porcine ESTs as possible be assembled into putative consensus transcripts. There are currently two sources of porcine EST assemblies: the Dana Farber Cancer Institute Porcine Gene Index (PGI) [originally housed at The Institute for Genome Research (TIGR); Quackenbush et al. 2000] and the Sino-Danish Pig Genome Project (SD) (Gorodkin et al. 2007). These two resources, however, do not fully exploit all available sequence data: PGI uses the public data within NCBI’s dbEST (Boguski et al. 1993) and dbCore, but not TRACE (Wheeler et al. 2008), which contains some sequences not found in dbEST or dbCore. Conversely, the SD assembly utilized the available public sequences at the time, which have more than doubled since their analysis was conducted. Other sequences databases, e.g., PEDE (Uenishi et al. 2007) and PiGenome (Lim et al. 2009), while useful for other purposes, do not provide comprehensive coverage of porcine ESTs.
Once the sequences are assembled, sequence similarity is improved due to longer sequences containing more structural information, such as multiple protein domains, than a single EST, which may contain only one or part of a protein domain. Hence, an assembled sequence can be used more reliably to leverage the better-annotated species for the less well-annotated species (Sjölander 2004). Homologies can be used not only to transfer annotation from one species to another, but also in cross-species comparisons to test whether the same structural homolog has similar characteristics such as expression pattern or protein function in different species, or if its character is unique to a given species.
The domestic pig, which has significant EST data and a developing genome sequence, has been used as a model for a number of investigations, including cystic fibrosis (Rogers et al. 2008) and tissue scarring (Gallant-Behm et al. 2007; Zhu et al. 2008). The pig genome has been modified for possible use in xenotransplantation (Lai and Prather 2002), but it does not have well-established annotations for most genes due to lack of a draft genome sequence. This makes it challenging to analyze the underlying pathways in such investigations. To fully utilize these models in large-scale transcriptomic studies, it is essential to create the best annotation of available ESTs.
Against this background, ANEXdb, an open-source web application that supports integrated access of two databases, ExpressDB and AnnotDB, that (1) house microarray expression and EST assembly and (2) annotation data, respectively, was created.
Materials and methods
ANEXdb application implementation
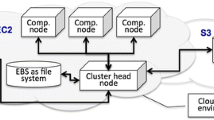
ANEXdb was implemented using a LAMP (Linux, Apache, MySQL, PHP/Perl) system, using Red Hat Enterprise Linux (RHEL) 4, Apache 2.0.53, MySQL5, and PHP5. The administrator uses Perl scripts to upload data into AnnotDB, to download the sequences via integration of NCBI’s eUtils (http://www.ncbi.nlm.nih.gov/entrez/query/static/eutils_help.html), and to control the assembly through the TGI Clustering tools (TGICL, Quackenbush et al. 2000, available at http://compbio.dfci.harvard.edu/tgi/software/). PHP scripts are used to control the data flow and analysis in ExpressDB via a Flash applet, a Java application for data migration, to upload data and run R procedures from the Bioconductor library for MAS/RMA calculations. A web-based query interface was created with PHP5 which allows users to query AnnotDB with a NCBI sequence ID, consensus ID, and Affymetrix probe ID, and experiment, hybridization, or sample information in the case of ExpressDB (Fig. 1).

Schema of the ANEXdb application. Arrows indicate data flow within ANEXdb; levels show which language or host is being used by ANEXdb to control data flow. A user has access to the submission and querying aspects of ANEXdb, while an administrator controls the finalization and migration of submission from a temporary database to the storage database; the administrator also controls the information going into AnnotDB
Currently, ExpressDB houses microarray expression data only from the Affymetrix platform. Support for Affymetrix was chosen since such data are available for many species, including pig, cattle, chicken, human, and mouse, and are also inherently comparable across experiments (Mongan et al. 2008). A submission interface implemented with PHP allows users to submit data from their microarray experiments to a temporary submission database. Such data include information about individual hybridizations such as sample information, i.e., organism and sample source, and technical information, i.e., hybridization and treatment protocols, and is MIAME compliant. An administrator (via an admin interface) verifies and approves the submission (a process called finalization). Once a submitted experiment is finalized, the system uses Bioconductor packages (Gentleman et al. 2004) to calculate MAS/RMA data for the experiment and transfers this and the previously submitted data to a final database that only an administrator can alter. The system can also output the data from a finalized experiment in the SOFT format for submission to GEO (Wheeler et al. 2008) by using a Java application that is invoked from the PHP interface. This application constructs an object model (an in-memory data representation) and uses the Apache Velocity template engine (http://velocity.apache.org/) to generate the SOFT output from the object model (Wheeler et al. 2008). The SOFT output and the.cel and.chp files are then zipped together into a single file that can be downloaded and submitted to GEO.
AnnotDB is designed to house individual or assemblies of sequences in a form that facilitates annotation of transcripts in the target species. Perl scripts were written to parse relevant data and to upload that data into AnnotDB. Such data include mappings of NCBI Gene database to GO terms, KEGG pathways, and the RefSeq database, as well as Pfam mappings to GO terms to help annotation of the sequences. To provide a basis for annotation, AnnotDB also includes several analyses done on the sequences from the assembly: sequence alignments (using BLAST and Exonerate), ORF prediction, and SNP analysis. To query AnnotDB, users use the same PHP interface that is used for ExpressDB. AnnotDB also has a MySQL guest account that allows users access to the complete database so they can perform database queries using Perl or another scripting language. This was not created for ExpressDB due to the potentially private nature of some of the data.
Sequence assembly
Currently within AnnotDB, 2,529,315 public porcine-expressed sequence entries have been assembled into consensus sequences (contigs). This assembly was compiled from NCBI’s dbEST 1,475,958 sequences, 18,157 sequences from dbCore, and 1,035,200 sequences from the TRACE Archive (February 2008). All sequences were downloaded in FASTA format and cleaned using the included SeqClean program (available at http://compbio.dfci.harvard.edu/tgi/software/) and two additional sequence files. One file was the sequences found in UniVec (Wheeler et al. 2008), while the other contained porcine-specific long and short repeated sequences created by querying Nucleotide at NCBI (available at http://www.anexdb.org/download.php). Using these two files, SeqClean functions removed or trimmed sequences that were vector sequences, regions of low complexity, repeated sequences, and any sequence less than 100 bases long. The removal of short sequences was performed twice: once prior to trimming the sequences, then after trimming poor regions off of good sequences. This resulted in 2,369,608 clean sequences, of which 1,144,310 were trimmed, that were available for assembly. These sequences were known to contain entries submitted to both dbEST and TRACE, but this partial duplication of sequences was retained in the initial assembly as duplicate entries in each database have differences in actual data submitted due to editing by submitters only to the dbEST pipeline.
The original FASTA annotation lines for each sequence were then mined for evidence that the sequence represented a full-length sequence by searching for “complete CDS” or “full length mRNA” keywords. Entries with such annotations were given the TGICL-based full-length “et|” designation in their annotation line and were then used for seeded clustering (see below).
Briefly, TGICL runs an alignment algorithm similar to megablast (Zhang et al. 2000) to calculate the overlap between each sequence using a given cutoff and then stores the alignment data in tab-delimited files. These files are then used to cluster the sequences into groups via a process called transitive clustering (tclust). Transitive clustering begins with the highest aligning pair and then groups other sequences that also align to either member of the pair. This is repeated with all sequences until none are left that align to any member in the cluster above the given threshold. An optional method, which requires sequences to be assigned the “et|” in their annotation line, is called seeded clustering (sclust). This starts with the “et|” sequences as a basis for a full-length and complete transcript and uses a stricter cutoff for sequences that overhang either end of the full-length sequence. We used sclust only on clusters with more than 20,000 individual sequence members. After the clustering, TGICL then uses CAP3 (Huang and Madan 1999) to assemble the clusters into contigs; it also performs a more stringent alignment that may cause a single cluster to create multiple contigs. Because TGICL calculates fewer overall alignments, this method is faster than the alignment and clustering procedure used by CAP3, which performs both at the same time.
Assembly annotation
The IPA was aligned against the RefSeq RNA database (Pruitt et al. 2007) using BLASTN (cutoff E value ≤ 1e-10, downloaded December 2008; Altschul et al. 2000), and against RefSeq Protein (cutoff E value ≤ 1e-5, downloaded February 2009) and Pfam (Bateman et al. 2002) databases (cutoff E value ≤ 1e-5, downloaded December 2008) using BLASTX. The IPA was also aligned with human chromosomes with Exonerate (est2genome model, requiring at least 60% of the sequence to be aligned, with at least one HSP with a minimum score of 100; Slater and Birney 2005). Using the resulting alignments to RefSeq, the RefSeq accession numbers were mapped to their NCBI Gene IDs (Wheeler et al. 2008) using the gene2refseq flat file (downloaded December 2008) from NCBI, which allowed the GO (Gene Ontology Consortium 2000) and KEGG (Kanehisa and Goto 2000) annotations to be transferred to the IPA from their homologs using gene2go (available from NCBI, downloaded December 2008). Likewise, the GO terms associated with each Pfam were transferred to the IPA using the pfam2go (downloaded December 2008) file from GO and the resulting alignments.
Similarly, to map the Exonerate alignments to the corresponding human gene, the gene2refseq file was used. This provided the chromosomal interval covered for each RefSeq ID and its corresponding Gene ID. These reported locations were then compared to the Exonerate alignments of the porcine sequences to the human chromosome. To allow for evolutionary variation in gene size, the human chromosomal interval for each RefSeq was extended 250 bp in each direction along the chromosome (an additional 500 bp). This overall interval was then compared with the interval along the human chromosome, which was aligned with the porcine sequence by using Exonerate, requiring the interval aligned with the porcine sequence to be within the extended human RefSeq interval. To validate the annotations, the Gene IDs of the top human hits transferred by the two RefSeq BLASTs (BLASTN and BLASTX) were compared with each other as well as the Gene IDs retrieved from Exonerate for each of the top hits.
Open reading frame (ORF) prediction
Using a Perl script, each sequence was submitted one at a time to the ORF Finder program (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the Standard genetic code. Perl scripting was used to extract all predicted reading frames in each of the six frames (if available) from the resulting output and inserted into the database. Open reading frames were predicted for all sequences in the entire IPA.
Array mapping
BLASTN was used to align both the Affymetrix porcine target and the individual 25-mer probe sequences to the entire IPA. A cutoff E value of ≤1e-5 was used for the target sequences, while, due to the shortness of the 25-mer, a cutoff E value of ≤0.05 was used for these sequences. For direct sequence comparisons between Affymetrix elements and IPA entities, the Affymetrix target sequences were blasted against the same RefSeq database at the same cutoff.
Assembly sequence variation and comparison to porcine dbSNP
To detect sequence variation from the assembly results, a Perl script was used to mine the ACE output file from CAP3 for differences between the consensus sequence and the sequences that contributed to each location along the consensus sequence. Because of the overlap between TRACE and dbEST for porcine sequences, frequencies of minor allele instances would be inaccurate if such duplicates were retained. Thus, a mapping of the two databases to identify unique entries was created either by matching the GenBank accession number when it was available in TRACE or by using the TRACE name to match against the description line within dbEST. This list was then filtered against the list of cleaned sequences from SeqClean. If both sequences were found to have remained after cleaning, the shorter of the two sequences was ignored during variant detection. Also, clusters that were reduced to a single unique read were completely ignored from sequence variation detection. Variant frequency was calculated using only the number of contributing sequences at the specific nucleotide location, not the overall number of contributing sequences for the entire consensus sequence.
To compare the SNPs identified from this data set to the porcine SNPs available in dbSNP at NCBI, the quarterly updated FASTA files were downloaded from dbSNP, joined, and then aligned using BLASTN with a match cutoff E value of ≤1e-5 (due to their short length and further criteria used; see below) to the ITCs. This created a subset of SNP-containing sequences from dbSNP that could be found in our assembly. These sequences from dbSNP were then mined to compare the exact location of the SNP within the submitted dbEST sequence to the corresponding base in the ITC. A percentage cutoff of the length of the dbSNP sequence in the alignment from 20 to 100% and the minimum number of minor allele reads were both utilized as further criteria in the analysis. The ITC accession number was then used to query the sequence variation table within the database to determine if that specific base was reported to contain a SNP or not. Also, since the ITCs are expressed sequences, but as dbSNP contains SNPs from both cDNA and genomic DNA sources, the FASTA annotation lines in dbEST were mined for the term “cDNA” to label SNPs specifically in expressed sequences. However, both genomic and cDNA clones were aligned and analyzed.
Results
Assembly and annotation of porcine-expressed sequences
Our assembly of all publicly available porcine-expressed sequences, called the Iowa Porcine Assembly (IPA), consists of 140,087 consensus sequences (contigs), called the Iowa Tentative Consensus (ITC), and 103,888 singletons. In comparison, the PGI contains 104,293 contigs and 133,455 singletons. A comparative analysis of the coverage of human RefSeqs by the IPA using BLAST showed that the IPA matched as many of the human sequences as did mouse RefSeqs: 72 vs. 73%, respectively (see Table 1 and Supplementary Fig. 1). For this BLAST analysis and all others in the following descriptions, we count a BLAST alignment for those results showing a BLASTN E value no larger than 1e-10 and a BLASTX E value no larger than 1e-05.
Of the 243,975 total sequences in the IPA, 191,602 (79%) have a BLASTN alignment to the RefSeq RNA database. The number of alignments to the RefSeq Protein and Pfam had a lower hit count: 71,332 (29%) and 76,386 (31%), respectively. However, the transfer of GO terms (from the two RefSeq databases and Pfam) and of KEGG terms (from the RefSeq databases) provided 166,119 and 92,263 sequences (68 and 38%), respectively, with additional functional information (see Table 2 for a summary of annotations).
The RefSeq annotations of the IPA sequences rely on the validity of the BLAST results between the IPA and RefSeq. To provide further evidence of these relationships, we compared the results of BLASTN and BLASTX hits to RefSeq with Exonerate alignments of the pig to the human genome sequence. We found that the majority of the top-scoring alignments overlapped: 80.1% of the top Exonerate alignments agreed with the top-scoring hits returned by at least one of the two BLAST algorithms. As well, 82% of the top BLASTX hits agreed with the top BLASTN hit, regardless of Exonerate results (see Fig. 2).

Highly consistent agreement of the top hits from the different alignment algorithms indicates IPA annotations are accurate. Chromosomal location of each Gene ID match from BLAST results was obtained using NCBI’s gene2refseq assignments and the top scoring RefSeq (with E value ≤ 1e-10 for BLASTN and E value ≤ 1e-5 for BLASTX alignments). The Gene ID location was then compared to the human genome location of the Exonerate alignment to the porcine sequence query; a match required the Exonerate-based alignment to be within 250 bases on either side of the BLAST Gene ID assignment. Note: numbers are different from Table 2 due to E-value cutoffs and because not all RefSeq IDs can be mapped to a Gene ID
A BLAST analysis of Affymetrix porcine GeneChip® target sequences to the IPA provided alignments for 22,569 of the 23,937 (94%) probe sets, of which 19,253 (80%) had an IPA to NCBI RefSeq alignment. This resulted in 1,293 (5.4%) more probe sets that aligned to RefSeq RNA than a direct BLASTN alignment of the Affymetrix target sequences to RefSeq RNA, which resulted in 17,960 alignments. The IPA-RefSeq alignments were also longer on average, which creates a higher score, with the IPA having an average BLASTN score of 1,244 versus an average score of 392 for the direct Affymetrix target sequence BLASTN against RefSeq.
Sequence variation inferred from overlapping sequences in AnnotDB
The ACE files containing the CAP3 assembly output for the ITCs were also analyzed for sequence variation. After removing duplicate sequences (totaling 747,939, see Materials and methods section), a total of 1,621,669 unique sequence reads were available between the three databases. Variation analysis yielded a maximum of 2,025,897 candidate SNPs, 12,264 candidate insertions, and 535,828 candidate deletions in 45,099 ITCs with putative variation using the minimum requirement of one minor allele variant. Although these numbers decreased as the number of minor allele reads required for declaration of variation increased, we were able to provide evidence for many SNPs; for example, nearly 30,000 SNPs were predicted in 3,141 different ITCs, even when ten sequences were required to show the minor allele variant (Table 3).
It is important to evaluate the accuracy of these predictions. We approached this by comparing our predictions for sequences in common with ITC sequences to available information in dbSNP. First, we identified those sequence entries in dbSNP that match any SNP-containing assembled contigs using BLAST. Then we determined whether the dbSNP entry has the exact SNP location as predicted in our assembly. By comparing dbSNP to our assembly (requiring a minimum of a 60% overlap length of the sequence from the dbSNP, and at least two minor allele reads), we were able to confirm that the majority of SNPs that were reported in regions in common between our predicted SNPs and those reported in dbSNP were cDNA derived (see Table 4 and Supplementary Fig. 2). Specifically, of the total number of porcine SNPs in dbSNP (34,508, including both genomic and cDNA entries), 18,235 had an alignment to an ITC, of which 9,919 (54%) were found to match putative sequence variation present in an ITC. However, when considering SNPs annotated as submitted from cDNAs to dbEST, this percentage rises to 98% (563 of 575 alignments) using the same criteria. When the criteria for declaring a putative SNP as matched to the public SNP data are reduced to the least stringent (only a single read of the minor allele is required, with any amount of sequence alignment), the percent of matches increases to 80% of the total porcine dbSNPs (16,125 of 20,095), and 99% (570 of 578) for the cDNA SNPs. Likewise, increasing the minimum number of minor alleles to three or increasing the minimum alignment length from 60 to 100% made little impact on these percentages for cDNA-derived SNPs. However, for SNPs submitted from a genomic sequence, increasing the minimum number of required alleles further reduces the percentage of SNPs in our assembly found in dbSNP. Like the cDNA-derived SNPs, no changes in the percentage match for genome-derived SNPs were observed by increasing the minimum cutoff of alignment (see Table 4 and Supplementary Fig. 2 for full results).
We also compared our SNP predictions to the available SD SNP data by using the SNP data reported on the SD website which requires at least three reads of the minor allele. We found a slightly lower number of ITC sequences with SNPs: 12,230 (ITC) versus those found by the SD consortium (14,120). However, there are more SNPs within the ITCs than in the SD consortium: 202,383(ITC) versus 54,828 (SD) (see Table 3).
Discussion
Assembly and annotation of porcine-expressed sequences
When our IPA is compared to the SD assembly (version 1, Gorodkin et al. 2007), which contains 48,629 contigs and 73,171 singletons, and to the DFCI Pig Gene Index assembly (PGI version 13, assembled July 2008), with 104,293 contigs and 133,455 singletons, the IPA has a higher number of contigs and fewer singletons. This is most likely due to our inclusion of more initial sequences than the SD consortium and to different assembly criteria than either of these two assemblies. In addition, unlike the SD assembly, we did not filter out ribosomal or mitochondrial mRNAs from the starting sequences. While we produced more contigs than what is currently thought to be the number of genes, this is expected due to the software distinguishing alternative splice products into distinct contigs or singletons. This has been documented for other assemblies; the human Gene Index at Dana Farber, for example, starts with more than seven million individual ESTs to create a Human Gene Index with over one million sequences. Similar results are seen with the mouse assembly. It is also likely many of these currently independent sequences will be merged once additional porcine sequence data become available.
The IPA covers as much of the human RefSeq database as the mouse RefSeq database (see Table 1 and Supplementary Fig. 1). This indicates that relative to human sequences, the IPA is as complete as the mouse RefSeq database. Also, due to the large percentage of the sequences in the IPA aligning to the entire RefSeq database (80%), putative gene annotation is provided to the majority of the IPA. By using Exonerate to map IPA sequences to the human chromosomes, it was also possible to get putative exon boundaries for 33,016 of the sequences in the IPA. This result will be updated when the porcine genome is complete and available. In addition to the direct sequence comparison to the RefSeq and Pfam databases, a large number of the sequences were annotated at varying levels by linking them to GO terms or KEGG pathways by using data from the Gene and Pfam databases. Thus, we believe this annotation is as complete as possible using available comparative information, and these data should be useful to researchers interested in annotation porcine sequences such as those obtained in transcriptional profiling experiments.
Array mapping to annotate probe set elements on the Affymetrix porcine GeneChip
An important goal of our work was to update the functional annotations of the porcine Affymetrix GeneChip probe sets. Through the use of homolog mapping to annotate Affymetrix GeneChip elements, it is also possible to obtain a better understanding of cross-species similarities and differences in transcriptional profiling experiments because the effects of structurally similar genes can be compared under similar experimental conditions. By aligning the Affymetrix target sequence to the IPA, and then the IPA to RefSeq, we obtained a higher average BLAST score than by aligning the Affymetrix target sequences directly to RefSeq. We also found more overall alignments.
Our method is similar to that reported by Tsai et al. (2006) but is different in a few ways. One is that we used only our consensus sequences to map to RefSeq entries instead of a mix of the Affymetrix target sequences and the PGI assembly while it was housed at TIGR (Quackenbush et al. 2000). We also used RefSeq instead of Ensembl (cDNAs are now a part of EMBL-Bank) because RefSeq is a more specific database than EMBL-Bank, containing well-annotated, nonredundant sequences (the NCBI equivalent of EMBL-Bank is GenBank). We also used a stricter cutoff: we set a cutoff E value of ≤1e-10, which translates into a minimum score of 78 in this situation, while Tsai et al. (2006) used a minimum score of 50. Finally, we mapped only the noncontrol porcine-specific sequences of the Affymetrix platform to the IPA, so we started with fewer probe sets (24,123 vs. 23,937). Due to our more stringent cutoff score, we did find slightly fewer alignments to RefSeq with the IPA sequences that have an Affymetrix sequence alignment than did their method: 19,253 (80%, IPA) vs. 19,675 (82%, Tsai et al.) at the comparative nucleotide level. Furthermore, 22,569 (94%) of the probe sets have an alignment to sequences in the IPA; thus, we can map annotations to Affymetrix probe sets that were recognized only when the longer and more complete ITC sequences were aligned with RefSeq. In addition, we provide online at AnnotDB a list of all the sequences within the IPA that each of the 25-mers hits to help flag possible cross-hybridization of close gene family members or alternate transcripts. Hence, by using RefSeq and a higher cutoff, we have greater confidence in the homologies being correct, and by leveraging all the additional information of newer sequences, we provide the most complete annotation of the Affymetrix platform for biological interpretation.
Sequence variation inferred from overlapping sequences in AnnotDB
Alignment of cDNA sequences from different individuals can provide evidence of structural variation, provided the depth of sequence data is sufficiently high. We found from 28,028 to 2,025,987 putative SNP variants, depending on the threshold for number of minor allele sequences required. A comparison between our assembly-based predictions and those SNPs reported within dbSNP entries, which were matched to our SNP-containing sequences, found a high frequency of dbSNP-reported variation in our SNP predictions, i.e., finding nearly all of those submitted to dbSNP originating from cDNA, and over half of those coming from genomic sequences that share a common region with an IPA (when requiring at least a 60% overlap length and two minor allele reads).
While altering the minimum number of minor alleles or the percentage of sequence coverage had no effect on matches to cDNA-based SNPs in dbSNP, altering the minimum minor allele number decreased the percentage of ITC SNPs found in dbSNP that were derived from genomic sequences. This decrease is due more to the decreased number of SNPs found in common from 15,505 (minimum minor allele count = 1) to 7,698 (minimum minor allele count = 3), when at least 60% of the dbSNP sequence is in the alignment than to a decrease in the number of alignments (from 19,010 to 17,148 for the respective minimum minor allele counts, see Table 4). This would indicate that the SNPs with the lowest minor allele count in the ITC data are not found in the population of dbSNP SNPs derived from genomic sequences with a minor allele count greater than one. This is not unexpected because the number of SNPs drops significantly as the minimum number of required minor allele reads increases, and thus dbSNP is queried with fewer SNPs.
These results provide evidence that many of our SNP predictions are accurate. However, this analysis could be biased because we could compare only a subset of our predicted SNPs to the SNPs reported in dbSNP, those with similarity to sequences that are already known to be polymorphic due to their inclusion in dbSNP. This could increase our rate of agreement; however, it is difficult to estimate the level of bias. Furthermore, we do identify in many cases the exact same polymorphic position as that reported in dbSNP for these matched sequences. Sequencing errors could be a source of false predictions. However, it would be very difficult to directly determine the false-positive rate of our predictions through resequencing of the source material; the public expressed sequence data set has more than two million sequences from animal samples from many contributing laboratories.
Another group has used sequence assembly to predict SNPs in porcine-expressed sequences (Panitz et al. 2007, see SD assembly information at http://pigest.ku.dk/download/pigest_kvl_1.0.1/snp_candidates.html). We found approximately fourfold more SNPs in the IPC than were found in the SD assembly at the same minor allele count. This large increase is most likely due to the inclusion of more sequences in the IPC than in the SD assembly and that additional sequences from breeds outside of those used in the SD project were included in the IPA. While the inclusion of additional sequences brings the possibility of increased incorrect pseudogene or close gene family member integration during the assembly process, artifacts created by assembling ESTs (Picoult-Newberg et al. 1999), and the inherent sequencing error rate from EST sequencing, full validation of the predicted SNPs in AnnotDB is beyond the scope of this article as such validation would require a draft porcine genome sequence to provide information on ITCs that may have been assembled incorrectly. However, it has been shown by others that approximately 40–74% of SNPs predicted by EST assembly were confirmed by resequencing or through Genetic Bit Analysis (see, e.g., Brett et al. 2000; Gorbach et al. 2009; Huntley et al. 2006; Panitz et al. 2007; Picoult-Newberg et al. 1999). Combining these results with the analysis above that shows substantial agreement with the known variations reported in dbSNP, we predict that the majority of predicted SNPs with at least three minor alleles (202,383; see Table 3) are likely to be accurate.
The porcine-specific sequence variation derived from these sequence assemblies can also be used in genome mapping or be combined with expression data to integrate changes in expression with the genome determinants of those changes (Cookson et al. 2009; Schadt 2006). For example, genome-wide SNP mapping using large-scale SNP panels are being used in many species, including the pig. Our SNP data are largely outside the randomly selected, genome sequencing-based SNPs in these panels and thus are complementary to these genotyping tools.
Availability, extensibility, and maintenance of annotation
By making ANEXdb open source, we provide a local installation option available to users in addition to the web-based tool. While ANEXdb can be a local installation for microarray data storage, it is GEO compatible due to its ability to output expression data in the SOFT format. Although ANEXdb is currently set up to output only in SOFT, the velocity template system will allow users to create custom outputs, such as XML-based MINiML, for submission to other databases such as ArrayExpress. Likewise, ANEXdb as an open-source application will also allow users to create novel plug-ins to directly access both expression and annotation data contained within the database. These user-created plug-ins can also be shared with other ANEXdb users.
We plan to reassemble the IPA and revise annotations approximately every 6 months and will incorporate the draft porcine genome sequence when it becomes available. Additional sequences will continue to help refine assemblies, and the genome sequence will provide an anchor to help determine alternative transcripts and misassembled highly similar gene family members. In addition to providing the new assembly and annotation data for online download, we will also store archived assembly and annotation data.
Although ANEXdb currently houses porcine-specific data, it has been designed to be species independent. ANEXdb can be easily customized for other species by populating the databases with the relevant annotation and expression data from a variety of platforms such as Affymetrix GeneChips in other species or custom arrays, including the new porcine 20 k spotted oligonucleotide array (Ernst et al. 2008). Finally, because ANEXdb holds both expressions and annotations in a single location, its use will allow easier and faster analysis of the large amounts of data generated through high-throughput expression experiments.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (2000) Basic local alignment search tool. J Mol Biol 215:403–410
Bateman A, Coin L, Durbin R, Finn RD, Hollich V et al (2002) The Pfam protein families database. Nucleic Acids Res 30:276–280
Boguski MS, Lowe TM, Tolstoshev CM (1993) dbEST—database for “expressed sequence tags”. Nat Genet 4:332–333
Brett D, Lehmann G, Hanke J, Gross S, Reich J et al (2000) EST analysis online: WWW tools for detection of SNPs and alternative splice forms. Trends Genet 16:416–418
Cookson W, Lian L, Abecasis G, Moffatt M, Lathrop M (2009) Mapping complex disease traits with global gene expression. Nat Rev Genet 10:184–194
Ernst CW, Steibel JP, Ramos AM, Lunney JK, Wysocki M et al (2008) Assessment of the swine protein-annotated oligonucleotide microarray and utility of the arrays for eQTL and transcriptional profiling studies. Plant and Animal Genome XVI, San Diego, CA, 12–16 January 2008, pp W494
Gallant-Behm CL, Reno C, Tsao H, Hart DA (2007) Genetic involvement in skin wound healing and scarring in domestic pigs: assessment of molecular expression patterns in (Yorkshire × Red Duroc) × Yorkshire backcross animals. J Invest Dermatol 127:233–244
Gene Ontology Consortium (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Gentleman R, Carey VJ, Bates DM, Bolstad B, Dettling M et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80
Gorbach DM, Hu ZL, Du ZQ, Rothschild MF (2009) SNP discovery in Litopenaeus vannamei with a new computational pipeline. Anim Genet 40:106–109
Gorodkin J, Cirera S, Hedegaard J, Gilchrist MJ, Panitz F et al (2007) Porcine transcriptome analysis based on 97 non-normalized cDNA libraries and assembly of 1,021,891 expressed sequence tags. Genome Biol 8:R45
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Huntley D, Baldo A, Johri S, Sergot M (2006) SEAN: SNP prediction and display program utilizing EST sequence clusters. Bioinformatics 22:495–496
Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30
Lai L, Prather RS (2002) Progress in producing knockout pigs for xenotransplantation by nuclear transfer. Ann Med 34:501–506
Lim D, Cho YM, Lee KT, Kang Y, Sung S et al (2009) The Pig Genome database (PiGenome): an integrated database for pig genome research. Mamm Genome 20:60–66
Mongan MA, Higgins M, Pine PS, Afshari C, Hamadeh H (2008) Assessment of repeated microarray experiments using mixed tissue RNA reference samples. Biotechniques 45:283–292
Nagaraj SH, Gasser RB, Ranganathan S (2006) A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief Bioinform 8:6–21
Panitz F, Stangaard H, Hornshøj H, Gorodkin J, Hedegaard J et al (2007) SNP mining porcine ESTs with MAVIANT, a novel tool for SNP evaluation and annotation. Bioinformatics 23:i387–i391
Picoult-Newberg L, Ideker TC, Pohl MG, Taylor SL, Donaldson MA et al (1999) Mining SNPs from EST databases. Genome Res 9:167–174
Pruitt KD, Tatusova T, Maglott DR (2007) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts, and proteins. Nucleic Acids Res 35:D61–D65
Quackenbush J, Cho J, Lee D, Liang F, Holt I et al (2000) The TIGR Gene Indices: reconstruction and representation of expressed gene sequences. Nucleic Acids Res 28:141–145
Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T et al (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 231:1837–1841
Schadt EE (2006) Novel integrative genomics strategies to identify genes for complex traits. Anim Genet 37:S18–S23
Sjölander K (2004) Phylogenomic inference of protein molecular function: advances and challenges. Bioinformatics 20:170–179
Slater G, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinform 6:31–41
Tsai S, Cassady JP, Freking A, Nonneman DJ, Rohrer GA et al (2006) Annotation of the Affymetrix porcine genome microarray. Anim Genet 37:423
Uenishi H, Eguchi T, Suzuki K, Sawazaki T, Toki D et al (2007) PEDE (Pig EST Data Explorer) has been expanded into Pig Expression Data Explorer, including 10,147 porcine full-length cDNA sequences. Nucleic Acids Res 35:D650–D653
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K et al (2008) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36:D13–D21
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Zhu KQ, Carrougher GJ, Couture OP, Tuggle CK, Gibran NS et al (2008) Expression of collagen genes in the cones of skin in the Duroc/Yorkshire porcine model of fibroproliferative scarring. J Burn Care Res 29:815–827
Acknowledgments
We thank the USDA CSREES-NRI-2005-3560415618 and the ISU Center for Integrated Animal Genomics for funding this project. A USDA MGET 2001-52100-11506 Fellowship to O.C. is gratefully acknowledged. KC was funded under a NIH-NSF BBSI-0234102 award to Iowa State University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Couture, O., Callenberg, K., Koul, N. et al. ANEXdb: an integrated animal ANnotation and microarray EXpression database. Mamm Genome 20, 768–777 (2009). https://doi.org/10.1007/s00335-009-9234-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-009-9234-1