
Abstract
Type 1 diabetes (T1D) is an autoimmune disease that has increased two- to threefold over the past half century by as yet unknown means. It is generally accepted that T1D is the result of gene–environment interactions, but such rapid increases in incidence are not explained by Mendelian inheritance. There have been numerous advances in our knowledge of the pathogenesis of T1D. Indeed, there has been a large number of genes identified that contribute to risk for this disease and several environmental factors have been proposed. The complexity of such interactions is yet to be understood for any major chronic disease. Epigenetic regulation is one way to explain the rapid increase in incidence and could be a central mechanism by which environmental factors influence development of diabetes. However, there is remarkably little known about the contribution of epigenetics to T1D pathogenesis. Here we speculate on various candidate processes and molecules of the immune and endocrine systems that could modify risk for T1D through epigenetic regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes (T1D) is a T-cell-mediated autoimmune disease that develops in genetically susceptible individuals whose immune system destroys the majority of insulin-secreting β cells in the pancreatic islets (Eizirik et al. 2009). The incidence of T1D has increased more than two- to threefold over the past half century, the most striking example being Finland where it has risen from approximately 12 to 63/100,000 (Knip and Siljander 2008; Patterson et al. 2009). This increase in incidence has not been paralleled by an increase in the frequency of the major risk genes, including MHC class II (DR, DQ), insulin, PTPN22, CTLA4, and IL-rRA (Barrett et al. 2009). Indeed, the prevalence of the classic MHC class II genes, which account for approximately 40% of genetic risk, appears to be decreasing (Fourlanos et al. 2008; Gillespie et al. 2004). There are now more than 40 risk loci associated with T1D, with the majority of non-MHC genes displaying odds ratios ≤1.2 (Barrett et al. 2009). Moreover, most individuals who possess T1D risk genes do not develop diabetes. Importantly, the concordance rate among monozygotic twins ranges from as low as 25 to 65% (Hyttinen et al. 2003; Redondo et al. 1999, 2008) and is approximately 6% in siblings. A common explanation has been that changes in the environment must be contributing to the increase in disease (Knip et al. 2005; Lefebvre et al. 2006). In particular, environmental exposures to dietary antigens and microbes have been implicated (Knip et al. 2005; Lefebvre et al. 2006). However, no single pathogenic environmental agent has been identified that explains all cases. In all likelihood, diabetes develops by various combinations of pathways in response to commonly encountered environmental exposures.
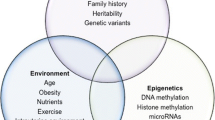
One way to address this complexity is to focus on central processes that control gene expression in response to environmental conditions. Epigenetic regulation is one such process by which mammals respond to environmental exposures (Jirtle and Skinner 2007). Although the complexity of the epigenome presents a formidable barrier to the study of its role in disease pathogenesis, a starting place could be to focus on epigenetic mechanisms that regulate candidate molecules and processes involved in β-cell development, survival, regeneration, and autoimmune reactions (Fig. 1). The purpose of this review is to highlight potential processes and targets that could be regulated epigenetically and help explain how environmental factors promote or inhibit the development of T1D.

Processes and genes potentially involved in environmental modification of T1D genetic risk. There are numerous risk genes for T1D, some of which could be modified by environmental exposures, the major factors being diet and microbes. The technology is becoming available to explore the epigenomes of selected molecules involved in T1D pathogenesis. The regulation of these key genes would be most evident in young high-risk individuals. It is difficult to access immune cells in the pancreatic islet infiltrate and the target β cells in human patients. Nonetheless, studies in peripheral blood mononuclear cells and pancreas sections from patients could reveal disease-specific epigenetic regulation of key genes. These targets could also be investigated in animal models of T1D
Epigenetic mechanisms
Epigenetics provides a mechanism by which environment can interact with identical genotypes to produce a variety of phenotypes. Although the DNA sequence is not changed, the phenotype is altered by epigenetic modifications of gene expression by mechanisms including methylation of DNA, post-translational modification of histones, or the activation of microRNAs (Wolffe and Matzke 1999). These modifications have the potential to be stable and heritable across cell divisions (Barski et al. 2007; Meissner et al. 2008; Wang et al. 2008). Changes to phenotype can be at the level of the cell, tissue, or whole organism.
The epigenome is modified in a number of different ways. The methylation of cytosine in CpG dinucleotides to form 5-methylcytosine has been well studied and is often associated with gene silencing (Bird 2002). Post-translational covalent histone modifications, including acetylation, methylation, phosphorylation, and ubiquitination, regulate gene silencing or expression (Bernstein et al. 2007). For example, lysine acetylation of histones H3 and H4 is commonly associated with an open chromatin conformation that allows for active gene expression, as does methylation of H3 lys4 (H3K4) and lys36 (H3K36), whereas H3 and H4 hypoacetylation and methylation of H3 lys9 (H3K9), H3 lys27 (H3K27), and H4 lys20 (H4K20) are associated with gene silencing (Bernstein et al. 2007). A third mechanism involves the expression of short noncoding RNAs, whose expression can lead to translational silencing through the specific binding and eventual degradation of transcribed RNA (Bernstein and Allis 2005). Despite considerable support for the involvement of environment in the expression of T1D (Knip et al. 2005; Lefebvre et al. 2006), evidence is scant regarding epigenetic mechanisms involved in T1D pathogenesis.
Epigenetics: a control mechanism for environmental modification of T1D?
The strongest evidence that environmental factors are important modifiers of T1D has been the lack of 100% concordance in twin pairs. Studies in monozygotic twins suggest that while genetics plays a significant role in the development of autoimmune type 1 diabetes, it is not sufficient to explain 35–75% of cases (Hyttinen et al. 2003; Redondo et al. 1999, 2008).
There is evidence that genetically identical twins can vary significantly in terms of DNA methylation content and histone modifications (Fraga et al. 2005). While 65% of monozygotic twin pairs demonstrated nearly identical 5-methylcytosine content in genomic DNA and H3 and H4 acetylation patterns, 35% of twin pairs did not. The differences correlated with age and time spent together; young twin pairs were more similar than older twin pairs, and those who had spent more time together were more similar than those who had been separated. Specific genomic locations were found to be differentially methylated, including expressed sequence tags or annotated single copy genes, and changes in the expression level of these genes correlated with the degree of CpG island methylation. While this study did not look at differences in disease susceptibility between twin pairs, it clearly demonstrated that the epigenotype of genetically identical individuals can diverge dramatically and that the extent of divergence is modified by environment and increases over time. This study provides one explanation for differences observed in time to disease onset and disease severity among genetically identical or similar individuals.
Epigenetic drift occurs due to small differences or defects in the transmission or maintenance of epigenetic information that accumulates through successive cell divisions (Bennett-Baker et al. 2003; Bjornsson et al. 2004; Cooney 1993; Jaenisch and Bird 2003). This can help to explain differences among tissues within an individual or between distant cells within a tissue (Yatabe et al. 2001). This likely influences the differences in epigenomes observed between twin pairs. The fidelity of maintenance of CpG methylation in mammalian cells has been estimated to be 97–99% per cell division (Ushijima et al. 2003). The error rate of methylation reactions has been estimated to be 2 × 10−5 per CpG site per day in the colon (Yatabe et al. 2001), a value that would change in a tissue-dependent manner. These errors create opportunities for changes in epigenetic profiles to occur and accumulate over time. Polymorphisms in genes that regulate the establishment or maintenance of methylation patterns can also impact the integrity of chromatin methylation patterns (Kanai and Hirohashi 2007), which can accumulate across generations of cells. In terms of T1D, pancreatic damage due to autoimmune attack leads to an increase in cellular proliferation (Kauri et al. 2007; Wang et al. 2005). Also, immune cells, including B and T lymphocytes involved in T1D autoimmunity, undergo rounds of proliferation and selection (Bergman and Cedar 2004). In both situations, the integrity of epigenetic marks that regulate appropriate damage repair or regeneration responses, insulin metabolism and regulation, or immune activation and maintenance could be altered and promote disease pathogenesis.
Epigenetics and immunity
T1D results from a T-cell-mediated autoimmune attack on pancreatic β cells. Epigenetics plays an important role in immune system development and function, specifically in T-cell maturation and cytokine gene expression. The differentiation of T-helper (TH) cells, which are protagonists in T1D autoimmunity, is subject to complex epigenetic control. There are three main lineages of TH cells: TH1, TH2, and TH17 (Harrington et al. 2005; Mosmann et al. 1986; Park et al. 2005). TH cells originate from naïve CD4+ T cells. External signals are transformed into specific cell-intrinsic changes, the end result of which is the differentiation of distinct TH cell lineages. The maturation of naïve T cells into TH cells represents a heritable transformation in that the cellular phenotype is passed on to daughter cells. While the genetic information encoded in the DNA sequence does not change, stable epigenetic modifications can be inherited (Barski et al. 2007; Meissner et al. 2008; Wang et al. 2008). The epigenetic control of T-cell differentiation was recently reviewed in detail (Aune et al. 2009; Sawalha 2008; Wilson et al. 2009). Here we present a brief general overview.
The differentiation of naïve T cells into TH cell lineages is driven by the expression of specific cytokines such as interferon (IFN)-γ, interleukin (IL)-4, and IL-17. A number of studies have indicated that modification of DNA methylation is a critical epigenetic process in TH-cell differentiation. As early as 1984, Ballas (1984) reported that treatment of thymoma cells with 5-azacytidine, which induces DNA hypomethylation, resulted in clones that constitutively produced IL-2, a key immunoregulatory cytokine involved in TH-cell differentiation. The data suggested that modifications to DNA methylation patterns could lead to changes in gene expression. It has also been reported that in the presence of 5-azacytidine, stimulated naïve TH cells showed increased IFN-γ and IL-4 expression (Bird et al. 1998), the main effector cytokines of TH1 and TH2 cells, respectively. Also, the Il4 locus is an area that undergoes extended and complex methylation modifications during TH-cell differentiation (Lee et al. 2002). A distinct 5′ region of the Il4 locus was hypermethylated in naïve TH cells, but during TH2-cell differentiation, this region was specifically demethylated. While demethylation was not an absolute necessity for IL-4 gene transcription, it was associated with strongly enhanced IL-4 gene expression in differentiated TH2 cells (Lee et al. 2002). Several other reports support the findings that changes in methylation of the Ifng and Il4 gene loci modulate the transcription of these key cytokines in TH1 and TH2 cells (Agarwal and Rao 1998; Jones and Chen 2006; Makar and Wilson 2004; Melvin et al. 1995; Santangelo et al. 2002). Therefore, the specificity and balance of cytokine expression in differentiating naïve TH cells is controlled at least in part at the level of the epigenome.
Of note, genomic hypomethylation of CD4+ T cells induces autoreactivity. CD4+ T cells, which normally do not react to self antigen, lose the requirement for specific antigen stimulation and are activated in response to self MHC class II molecules when treated with 5-azacytidine (Richardson 1986). The ability of the T cells to mount an autoimmune response is due to increased expression of the adhesion molecule lymphocyte function-associated antigen (LFA)-1. LFA-1 is involved in the stabilization of the T-cell receptor (TCR)–MHC interaction and provides costimulatory signals for the activation of T-cell responses (Wulfing et al. 2002). The change in its expression is the result of demethylation of regulatory regions in the 5′ promoter of ITGAL, a subunit of LFA-1, and subsequent loss of its transcriptional inhibition (Lu et al. 2002; Richardson et al. 1992). Therefore, the overexpression of LFA-1 is hypothesized to stabilize lower-affinity TCR–MHC interactions and provide costimulation, which leads to an autoimmune response resembling that of systemic lupus erythematosus. Together, the data indicate that changes in the regulation of methylation-sensitive genes can lead to the promotion of an autoimmune response by allowing indiscriminate interactions between T cells and antigen-presenting cells. This could also play a role in β-cell autoimmunity.
Post-translational histone modifications have also been shown to selectively control gene transcription in specific T-cell populations. In a recent study, Wei et al. (2009) performed global genome mapping of histone H3K4 and H3K27 trimethylation (me3) in naïve T, TH, and T regulatory (Treg) cells using a ChIP-Seq high-throughput DNA sequencing approach. The goal was to determine whether the changes associated with terminal differentiation of specific T-cell populations were reflected in heritable epigenetic modifications. They found that the H3K27me3 histone modification was associated with gene suppression and was detected in the Il4 gene in naïve T, TH1, and TH17 cells. The study also revealed that genes encoding T-cell lineage-specific transcription factors were targeted by particular epigenetic modifications. Transcription factors have been shown to bind a region within the Ifng locus of TH1 and TH2 cells that is associated with histone modifications characteristic of relaxed, accessible chromatin (Hatton et al. 2006). In TH1 cells, the Ifng locus showed selective T-bet binding and activity, a key transcription factor associated with TH1 proinflammatory responses (Peng 2006).
Epigenetic modifications also play a role in the maturation and function of Treg cells, which are essential players in the maintenance of self-tolerance (Lal et al. 2009). Foxp3 is the main transcription factor that drives natural Treg-cell differentiation (Sgouroudis and Piccirillo 2009). The loss of Foxp3 in mice results in the failure to generate CD25+CD4+ Treg cells and the development of severe autoimmunity (Fontenot et al. 2003). Foxp3 expression in Treg cells has been shown to be epigenetically regulated. Using bisulfite sequencing and ChIP, Floess et al. (2007) showed that a conserved and transcriptionally active region within the Foxp3 locus was completely demethylated at CpG motifs in Foxp3+CD25+CD4+ Treg cells, but not in naïve CD25-CD4+ T cells. H3 acetylation and trimethylation patterns indicative of open euchromatin were observed in this region and were also associated with Treg differentiation (Floess et al. 2007). Kim and Leonard (2007) reported another CpG region within the Foxp3 locus, the methylation state of which inversely correlated with Foxp3 expression. Several other studies have also indicated that DNA methylation plays a role in the Foxp3 gene expression in mouse and human Treg cells (Janson et al. 2008; Lal et al. 2009). The normal function of Treg cells is to promote tolerance and prevent autoimmunity and therefore it has been suggested that their dysregulation plays a role in diabetes pathogenesis (Sgouroudis and Piccirillo 2009).
The autoimmune process observed in T1D is driven by an inflammatory response to self antigens. One of the main effector transcription factors involved in this process is NF-κB. The expression of a number of inflammation-associated cytokines is dependent on activation by NF-κB. Activated NF-κB enters the nucleus, but it requires remodeling of the chromatin to induce gene transcription (Natoli et al. 2005). Inflammatory stimuli have been shown to induce post-translational modifications of the chromatin at specific NF-κB-regulated cytokine or chemokine genes, including IL-8 and macrophage chemoattractant protein-1, followed by the recruitment of NF-κB to their promoters. This includes increased H4 acetylation (Saccani et al. 2001), phosphorylation, and phosphoacetylation of H3 (Saccani et al. 2002) and altered H3K9 methylation (Saccani and Natoli 2002). It will be necessary to determine whether T1D-associated inflammation also mediates gene expression changes through the modification of chromatin structure.
In their application of the ChIP-chip approach to study immune cells from T1D patients and control subjects, Miao et al. (2008) observed that lymphocytes from T1D patients demonstrated specific histone modification patterns. H3K9 dimethylation (me2) at the promoters of a number of genes associated with inflammation and autoimmunity, including CTLA4, a T1D susceptibility gene, and TGF-β, NF-κB, p38 mitogen-activated protein kinase, Toll-like receptors, and IL-6, was observed in patients but not controls. An important caveat in these studies is that in earlier findings from the same group, human THP-1 monocytes cultured in high-glucose conditions also revealed alterations in histone lysine methylation of various T1D-related genes (Miao et al. 2007). Because blood glucose levels vary considerably in patients with T1D, these authors point out that it is difficult to differentiate whether such changes are related to diabetes pathogenesis or to variations in blood glucose (Miao et al. 2008). Nonetheless, these data suggest that the genes were tagged for active transcription and could therefore influence disease outcome through changes in gene expression.
Epigenetic modifications in the target β cells
Specific gene expression profiles are important for the proper development, function, and repair response of pancreatic β cells. Both type 1 and type 2 diabetes are associated with progressive dysfunction of insulin-secreting β cells, including dysregulated expression of genes involved in β-cell development and function (Eizirik et al. 2009; Matveyenko and Butler 2008). Insulin gene expression has been shown to be dependent on specific patterns of histone acetylation and methylation. Specifically, the proximal promoter of the Ins1/2 gene in pancreatic β cells displays H3 hyperacetylation and hypermethylation at H3K4, both of which are associated with euchromatin status and active gene transcription (Chakrabarti et al. 2003; Mutskov et al. 2007). Dimethylation status at H3K4 in the regulatory region of Ins1/2 was shown to be maintained by a transcriptional protein complex involving Pdx1, a key transcription factor involved in β-cell development (Babu et al. 2007), and the methyltransferase Set7/9, which promoted the continuous activation of Ins1/2 and other genes involved in glucose-stimulated insulin secretion (Deering et al. 2009; Francis et al. 2005). Therefore, changes to histone modifications associated with the insulin gene could alter the function of β cells by influencing replication or expression of autoantigens on their surface, thereby modifying diabetes pathogenesis.
Impaired insulin and glucose metabolism in T1D alters methyl group metabolism
In contrast to the previously described epigenetic changes that could be involved in the initiation or progression of autoimmune T1D, the diabetic physiological state can also modify cellular methylation potential by altering methyl group metabolism. Homocysteine and methionine metabolism are interdependent and regulate the availability of methyl groups for cellular methylation reactions (Fig. 2) (Fox and Stover 2008). Homocysteine can be remethylated to form methionine, which in turn can be converted to S-adenosylmethionine (AdoMet). AdoMet is the major methyl group donor in cellular methylation reactions, including the methylation of DNA, histones, and other molecules (Fox and Stover 2008).

Methionine synthesis cycle. Cellular methylation potential is dependent on the production of S-adenoyslmethionine (AdoMet), the major methyl donor in the cell. Methionine is generated when homocysteine is remethylated. A methyl group can be transferred from 5-methyltetrahydrofolate (THF), which is catalyzed by the B12-dependent enzyme methionine synthase (MS). 5-MethylTHF is produced by methyleneTHF reductase (MTHFR) from the folate metabolic pathway. In the liver and kidney, homocysteine can be remethylated by betaine:homocysteine methyltransferase (BHMT), a reaction that depends on choline-derived betaine as the methyl donor. Methionine is converted to AdoMet, from which methyltransferases (MT) transfer methyl groups to acceptor molecules. The reaction produces S-adenosylhomocysteine (AdoHcy) that can be converted to homocysteine. Homocysteine can be metabolized by the B6-dependent transsulfuration pathway. SHMT1, cytoplasmic serine hydroxymethyltransferase
Insulin concentrations have been shown to correlate positively with homocysteine (Chiang et al. 2009; Fonseca et al. 2000). In autoimmune T1D, patients demonstrate lower rates of homocysteine remethylation and methionine synthesis and increased rates of homocysteine transsulfuration, changes that are normalized by insulin treatment (Abu-Lebdeh et al. 2006). Similarly, in rats with streptozotocin-induced diabetes, metabolism of homocysteine by transsulfuration is increased, as indicated by an increase in transsulfuration pathway enzymes, which can also be normalized by insulin treatment (Jacobs et al. 1998; Ratnam et al. 2002). In fact, insulin treatment of rats with streptozotocin-induced diabetes decreases the activities of transsulfuration enzymes resulting in a dose-dependent increase in plasma homocysteine (Gursu et al. 2002). Insulin has also been shown to induce folate-dependent homocysteine remethylation pathways under methionine-restricted conditions in hepatic cells (Chiang et al. 2009). Together the data suggest that insulin increases homocysteine levels and homocysteine remethylation while simultaneously reducing the capacity to eliminate homocysteine by transsulfuration.
Glucose has also been shown to regulate homocysteine metabolism. Homocysteine has been shown to be inversely correlated with blood glucose concentrations in type 2 diabetic patients (Mazza et al. 2005). It has also been demonstrated that glucose concentration is negatively correlated with the activity of the enzyme methylenetetrahydrofolate reductase, which provides methyl groups for the remethylation of homocysteine (Fig. 2) (Dicker-Brown et al. 2001). Glucose is also positively correlated with the activity of enzymes involved in homocysteine transsulfuration in cultured hepatocytes (Dicker-Brown et al. 2001). The data suggest that changes in homocysteine are due to its metabolism by the transsulfuration pathway. However, Chiang et al. (2009) showed that homocysteine clearance under high-glucose conditions in cultured hepatocytes could in fact be due to increased homocysteine remethylation flux. Interestingly, these authors also found that high glucose levels were associated with increased cellular methylation potential, as indicated by the AdoMet-to-S-adenosylhomocysteine (AdoHcy) ratio, increased AdoMet synthase activity, and DNA methyltransferase activity and global methylation potential in cultured hepatocytes (Chiang et al. 2009). In addition, glucose has recently been shown to modify chromatin structure and influence global gene transcription control (Rathmell and Newgard 2009; Wellen et al. 2009). The diabetic state is characterized by fluctuations in insulin and glucose, which clearly influence homocysteine and methionine metabolism. This has the potential to alter cellular methylation reactions in various tissues and could be related to diabetes-associated complications. It also poses a problem for the interpretation of studies of epigenetic modifications in patients with T1D whose blood glucose levels often display wide variation. It will be important to study high-risk, normoglycemic individuals as they progress to islet autoimmunity (i.e., presence of autoantibodies), before the appearance of overt diabetes.
Modification of epigenetics by diet-derived methyl cofactors
The establishment and maintenance of methylation patterns of CpG dinucleotides in DNA and histones depend on cellular methylation potential and one-carbon (methyl group) metabolism, which is itself dependent on various nutrients. Folate, the biologically active form of folic acid, mediates one-carbon metabolism (Fig. 2) (Fox and Stover 2008). A major product of one-carbon metabolism is methionine, which can be converted to AdoMet, the main methyl donor in cellular methylation reactions (Fox and Stover 2008). Methionine synthase, a vitamin B12-dependent enzyme, transfers a methyl group from 5-methyltetrahydrofolate to homocysteine to form methionine (Fig. 2). Homocysteine metabolism by the transsulfuration pathway requires vitamin B6. The B6-dependent enzyme serine hydroxymethyltransferase is a source of 5,10-methylenetetrahydrofolate, which is the substrate required for folate-dependent methionine synthesis. Betaine, which can be taken up from the diet or formed from choline, can also be used for homocysteine remethylation and methionine synthesis in liver and kidney (Zeisel 2006). Other nutrients that influence one-carbon metabolism include niacin, riboflavin, zinc, cobalt, and vitamin A (Fox and Stover 2008). Therefore, nutrient deficiencies or single nucleotide polymorphisms in enzymes involved in methyl group metabolism can influence methyl group availability as well as the establishment and maintenance of epigenetic marks.
Maternal and early postnatal diet plays a significant role in the establishment of the epigenome in the developing offspring, which can be maintained throughout its lifetime (Waterland and Michels 2007). The epigenome is likely the most plastic and sensitive to environmental-mediated changes during embryonic development. Shortly after fertilization, gamete-related epigenetic marks are removed and replaced with embryonic marks (Morgan et al. 2005). The developing gametes also undergo epigenetic reprogramming in which the parental marks are replaced by stem cell marks (Morgan et al. 2005). It is during these periods of flux that environmental factors are most likely to contribute to epigenetic diversity. Jirtle and his group have used the Agouti mouse model to clearly demonstrate the principle that maternal nutritional status or exposure to other environmental factors, including toxins, alters genomic methylation profiles with unknown but potentially long-term, heritable and possibly detrimental outcomes for the offspring (Dolinoy et al. 2007; Waterland and Jirtle 2003).
It is well known that early diet affects the development of diabetes in humans (Norris et al. 2003; Ziegler et al. 2003), as well as in diabetes-prone BB rats and NOD mice (Lefebvre et al. 2006). Whether the various dietary manipulations occur via epigenetic regulation remains a matter of speculation. It is known that changes in maternal diet can be associated with the abnormal development of the pancreas in the offspring. Feeding of a low-protein diet to NOD mice during pregnancy and until the offspring were weaned incurred effects on both islet mass (decreased) and autoimmunity (inflammatory cytokines decreased) (Chamson-Reig et al. 2009). It is also known that feeding a low-protein diet to rat dams decreased islet mass and several measures of islet vascularity (Boujendar et al. 2003). These effects were reversed by feeding a taurine supplement and it has also been reported that taurine supplements can delay diabetes onset in NOD mice (Arany et al. 2004). It has been speculated, though not yet proven, that such changes observed in offspring are due to epigenetic modifications in the developing embryo. However, more studies will be required to determine the mechanisms involved and how they influence chronic diseases such as T1D.
Conclusions
The major risk genes for T1D have been known for decades, but the cause of T1D remains unclear. Part of the problem may lie in the fact that there are numerous pathways through which diabetes can develop in different individuals. The layers of genetic complexity combined with the role of different environmental exposures occurring at different ages and at varying doses present a formidable problem. If the epigenome is a key controller of gene–environment interactions as some suspect, then it will be important to understand epigenetic regulation of key T1D-related genes and how this leads to the development of diabetes.
The epigenome is a conduit by which the environment can influence the expression of the genome and ultimately phenotype. We have speculated that epigenetics can influence T1D outcome in a number of ways. β-cell development, maintenance, metabolism, and regeneration can all be influenced by epigenetic mechanisms. Also, immune responses, including the activation of T cells and induction of Treg cells, rely on appropriate epigenetic regulation. Furthermore, insulin and glucose metabolism influence the epigenome of tissues such as the liver, and potentially the pancreas, which could contribute to T1D-associated pathologies. Clearly, information on the epigenetic mechanisms at play in the development of T1D is extremely limited and new studies are required. The hope is that this information will provide new understanding of the pathogenesis, potential treatments, or even prevention of T1D.
References
Abu-Lebdeh HS, Barazzoni R, Meek SE, Bigelow ML, Persson XM et al (2006) Effects of insulin deprivation and treatment on homocysteine metabolism in people with type 1 diabetes. J Clin Endocrinol Metab 91:3344–3348
Agarwal S, Rao A (1998) Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9:765–775
Arany E, Strutt B, Romanus P, Remacle C, Reusens B et al (2004) Taurine supplement in early life altered islet morphology, decreased insulitis and delayed the onset of diabetes in non-obese diabetic mice. Diabetologia 47:1831–1837
Aune TM, Collins PL, Chang S (2009) Epigenetics and T helper 1 differentiation. Immunology 126:299–305
Babu DA, Deering TG, Mirmira RG (2007) A feat of metabolic proportions: Pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis. Mol Genet Metab 92:43–55
Ballas ZK (1984) The use of 5-azacytidine to establish constitutive interleukin 2-producing clones of the EL4 thymoma. J Immunol 133:7–9
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD et al (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41:703–707
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Bennett-Baker PE, Wilkowski J, Burke DT (2003) Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165:2055–2062
Bergman Y, Cedar H (2004) A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat Rev Immunol 4:753–761
Bernstein E, Allis CD (2005) RNA meets chromatin. Genes Dev 19:1635–1655
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128:669–681
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA et al (1998) Helper T cell differentiation is controlled by the cell cycle. Immunity 9:229–237
Bjornsson HT, Cui H, Gius D, Fallin MD, Feinberg AP (2004) The new field of epigenomics: implications for cancer and other common disease research. Cold Spring Harb Symp Quant Biol 69:447–456
Boujendar S, Arany E, Hill D, Remacle C, Reusens B (2003) Taurine supplementation of a low protein diet fed to rat dams normalizes the vascularization of the fetal endocrine pancreas. J Nutr 133:2820–2825
Chakrabarti SK, Francis J, Ziesmann SM, Garmey JC, Mirmira RG (2003) Covalent histone modifications underlie the developmental regulation of insulin gene transcription in pancreatic beta cells. J Biol Chem 278:23617–23623
Chamson-Reig A, Arany EJ, Summers K, Hill DJ (2009) A low protein diet in early life delays the onset of diabetes in the non-obese diabetic mouse. J Endocrinol 201:231–239
Chiang EP, Wang YC, Chen WW, Tang FY (2009) Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J Clin Endocrinol Metab 94:1017–1025
Cooney CA (1993) Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging 57:261–273
Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG (2009) Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes 58:185–193
Dicker-Brown A, Fonseca VA, Fink LM, Kern PA (2001) The effect of glucose and insulin on the activity of methylene tetrahydrofolate reductase and cystathionine-beta-synthase: studies in hepatocytes. Atherosclerosis 158:297–301
Dolinoy DC, Huang D, Jirtle RL (2007) Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104:13056–13061
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5:219–226
Floess S, Freyer J, Siewert C, Baron U, Olek S et al (2007) Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 5:e38
Fonseca V, Dicker-Brown A, Ranganathan S, Song W, Barnard RJ et al (2000) Effects of a high-fat-sucrose diet on enzymes in homocysteine metabolism in the rat. Metabolism 49:736–741
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4:330–336
Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC et al (2008) The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care 31:1546–1549
Fox JT, Stover PJ (2008) Folate-mediated one-carbon metabolism. Vitam Horm 79:1–44
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102:10604–10609
Francis J, Chakrabarti SK, Garmey JC, Mirmira RG (2005) Pdx-1 links histone H3-Lys-4 methylation to RNA polymerase II elongation during activation of insulin transcription. J Biol Chem 280:36244–36253
Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR et al (2004) The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet 364:1699–1700
Gursu MF, Baydas G, Cikim G, Canatan H (2002) Insulin increases homocysteine levels in a dose-dependent manner in diabetic rats. Arch Med Res 33:305–307
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL et al (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132
Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM et al (2006) A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25:717–729
Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J (2003) Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes 52:1052–1055
Jacobs RL, House JD, Brosnan ME, Brosnan JT (1998) Effects of streptozotocin-induced diabetes and of insulin treatment on homocysteine metabolism in the rat. Diabetes 47:1967–1970
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254
Janson PC, Winerdal ME, Marits P, Thorn M, Ohlsson R et al (2008) FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS ONE 3:e1612
Jirtle RL, Skinner MK (2007) Environmental epigenomics and disease susceptibility. Nat Rev Genet 8:253–262
Jones B, Chen J (2006) Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J 25:2443–2452
Kanai Y, Hirohashi S (2007) Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis 28:2434–2442
Kauri LM, Wang GS, Patrick C, Bareggi M, Hill DJ et al (2007) Increased islet neogenesis without increased islet mass precedes autoimmune attack in diabetes-prone rats. Lab Invest 87:1240–1251
Kim HP, Leonard WJ (2007) CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med 204:1543–1551
Knip M, Siljander H (2008) Autoimmune mechanisms in type 1 diabetes. Autoimmun Rev 7:550–557
Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O et al (2005) Environmental triggers and determinants of type 1 diabetes. Diabetes 54(Suppl 2):S125–S136
Lal G, Zhang N, van der Touw W, Ding Y, Ju W et al (2009) Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol 182:259–273
Lee DU, Agarwal S, Rao A (2002) Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 16:649–660
Lefebvre DE, Powell KL, Strom A, Scott FW (2006) Dietary proteins as environmental modifiers of type 1 diabetes mellitus. Annu Rev Nutr 26:175–202
Lu Q, Ray D, Gutsch D, Richardson B (2002) Effect of DNA methylation and chromatin structure on ITGAL expression. Blood 99:4503–4508
Makar KW, Wilson CB (2004) DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol 173:4402–4406
Matveyenko AV, Butler PC (2008) Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab 10(Suppl 4):23–31
Mazza A, Bossone E, Mazza F, Distante A (2005) Reduced serum homocysteine levels in type 2 diabetes. Nutr Metab Cardiovasc Dis 15:118–124
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J et al (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770
Melvin AJ, McGurn ME, Bort SJ, Gibson C, Lewis DB (1995) Hypomethylation of the interferon-gamma gene correlates with its expression by primary T-lineage cells. Eur J Immunol 25:426–430
Miao F, Wu X, Zhang L, Yuan YC, Riggs AD et al (2007) Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem 282:13854–13863
Miao F, Smith DD, Zhang L, Min A, Feng W et al (2008) Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 57:3189–3198
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14 Spec No 1:R47–R58
Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL (1986) Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136:2348–2357
Mutskov V, Raaka BM, Felsenfeld G, Gershengorn MC (2007) The human insulin gene displays transcriptionally active epigenetic marks in islet-derived mesenchymal precursor cells in the absence of insulin expression. Stem Cells 25:3223–3233
Natoli G, Saccani S, Bosisio D, Marazzi I (2005) Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol 6:439–445
Norris JM, Barriga K, Klingensmith G, Hoffman M, Eisenbarth GS et al (2003) Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA 290:1713–1720
Park H, Li Z, Yang XO, Chang SH, Nurieva R et al (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, EURODIAB Study Group (2009) Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet 373:2027–2033
Peng SL (2006) The T-box transcription factor T-bet in immunity and autoimmunity. Cell Mol Immunol 3:87–95
Rathmell JC, Newgard CB (2009) Biochemistry. A glucose-to-gene link. Science 324:1021–1022
Ratnam S, Maclean KN, Jacobs RL, Brosnan ME, Kraus JP et al (2002) Hormonal regulation of cystathionine beta-synthase expression in liver. J Biol Chem 277:42912–42918
Redondo MJ, Rewers M, Yu L, Garg S, Pilcher CC et al (1999) Genetic determination of islet cell autoimmunity in monozygotic twin, dizygotic twin, and non-twin siblings of patients with type 1 diabetes: prospective twin study. BMJ 318:698–702
Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T (2008) Concordance for islet autoimmunity among monozygotic twins. N Engl J Med 359:2849–2850
Richardson B (1986) Effect of an inhibitor of DNA methylation on T cells. II. 5-Azacytidine induces self-reactivity in antigen-specific T4+ cells. Hum Immunol 17:456–470
Richardson BC, Strahler JR, Pivirotto TS, Quddus J, Bayliss GE et al (1992) Phenotypic and functional similarities between 5-azacytidine-treated T cells and a T cell subset in patients with active systemic lupus erythematosus. Arthritis Rheum 35:647–662
Saccani S, Natoli G (2002) Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 16:2219–2224
Saccani S, Pantano S, Natoli G (2001) Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med 193:1351–1359
Saccani S, Pantano S, Natoli G (2002) p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3:69–75
Santangelo S, Cousins DJ, Winkelmann NE, Staynov DZ (2002) DNA methylation changes at human Th2 cytokine genes coincide with DNase I hypersensitive site formation during CD4(+) T cell differentiation. J Immunol 169:1893–1903
Sawalha AH (2008) Epigenetics and T-cell immunity. Autoimmunity 41:245–252
Sgouroudis E, Piccirillo CA (2009) Control of type 1 diabetes by CD4+Foxp3+ regulatory T cells: lessons from mouse models and implications for human disease. Diabetes Metab Res Rev 25:208–218
Ushijima T, Watanabe N, Okochi E, Kaneda A, Sugimura T et al (2003) Fidelity of the methylation pattern and its variation in the genome. Genome Res 13:868–874
Wang GS, Rosenberg L, Scott FW (2005) Tubular complexes as a source for islet neogenesis in the pancreas of diabetes-prone BB rats. Lab Invest 85:675–688
Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A et al (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40:897–903
Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23:5293–5300
Waterland RA, Michels KB (2007) Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 27:363–388
Wei G, Wei L, Zhu J, Zang C, Hu-Li J et al (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30:155–167
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR et al (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080
Wilson CB, Rowell E, Sekimata M (2009) Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 9:91–105
Wolffe AP, Matzke MA (1999) Epigenetics: regulation through repression. Science 286:481–486
Wulfing C, Sumen C, Sjaastad MD, Wu LC, Dustin ML et al (2002) Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat Immunol 3:42–47
Yatabe Y, Tavare S, Shibata D (2001) Investigating stem cells in human colon by using methylation patterns. Proc Natl Acad Sci U S A 98:10839–10844
Zeisel SH (2006) Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr 26:229–250
Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E (2003) Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 290:1721–1728
Acknowledgments
Studies in the laboratory of FWS have been supported by the Juvenile Diabetes Research Foundation, Canadian Institutes of Health Research, and Canadian Diabetes Association. AJM is supported by Health Canada. AS is a Juvenile Diabetes Research Foundation Postdoctoral Fellow.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
MacFarlane, A.J., Strom, A. & Scott, F.W. Epigenetics: deciphering how environmental factors may modify autoimmune type 1 diabetes. Mamm Genome 20, 624–632 (2009). https://doi.org/10.1007/s00335-009-9213-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-009-9213-6