
Abstract
A quantitative trait locus (QTL) was identified by linkage analysis on bovine Chromosome 19 that affects the fatty acid, myristic acid (C14:0), in subcutaneous adipose tissue of pasture-fed beef cattle (99% level: experiment-wise significance). The QTL was also shown to have significant effects on ten fatty acids in the milk fat of pasture-fed dairy cattle. A positional candidate gene for this QTL was identified as fatty acid synthase (FASN), which is a multifunctional enzyme with a central role in the metabolism of lipids. Five single nucleotide polymorphisms (SNPs) were identified in the bovine FASN gene, and animals were genotyped for FASN SNPs in three different cattle resource populations. Linkage and association mapping results using these SNPs were consistent with FASN being the gene underlying the QTL. SNP substitution effects for C14:0 percentage were found to have an effect in the opposite direction in adipose fat to that in milk fat. It is concluded that SNPs in the bovine FASN gene are associated with variation in the fatty acid composition of adipose fat and milk fat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dietary fatty acids are known to have a major influence on human health, particularly in countries whose populations are well-nourished, such as in developed countries where milk and meat are major components of dietary intake. It is generally accepted that saturated fats containing fatty acids with a chain length of 14 or 16 carbons, commonly found in beef meat and cow’s milk, are risk factors in coronary heart disease (Mozaffarian et al. 2005). For example, myristic acid (C14:0) is hypercholesterolemic and raises concentrations of both low-density lipoprotein and high-density lipoprotein cholesterol compared with oleic acid (C18:1) (Temme et al. 1997). In contrast, those fatty acids with a carbon chain length of at least 18, especially unsaturated fatty acids, are considered to be beneficial to human health.
Methods are available to modify fatty acid composition in animal products that contain oil or fat, including chemical extraction, blending, and also specific feeding regimens for cattle (Baumgard et al. 2005). However, such methods are likely to incur a significant extra cost in producing these animal fats, which are relatively low-value commodity products. The methods may also compromise flavor, mouth feel, or other physical properties of the animal products (Berner 1993). In contrast to the farm livestock industry, the plant industry has been very successful in manipulating or tailoring fatty acid composition, especially in seed oil, via breeding or transgenics to improve edible traits and oils for industrial use (Davies 1996; Singh et al. 2005). Is a genetic approach to manipulating fatty acid composition feasible in livestock as well?
There have been many reports of quantitative trait loci (QTL) in farm livestock that affect fat deposition, but there are only a few QTLs known to affect fatty acid composition. A QTL on porcine Chromosome 4 that affects the proportions of linoleic and oleic acids has been described; individual pigs homozygous for the allele from the Iberian breed had 1.5% less linoleic acid, on average, than those homozygous for the Landrace allele (Perez-Enciso et al. 2000). A later study (Clop et al. 2003) also revealed QTL for a number of traits related to fatty acid composition, including a fatty acid double bond index, an unsaturated fat index, and percentages of linolenic, gadoleic, and myristic acids. Other investigators (Lee et al. 2003) identified two QTLs on porcine Chromosomes 1 and 18 that affected fatty acid composition (linoleic and myristic acids, respectively). A recent study in sheep has identified significant QTLs at the genome-wide level for fatty acid composition in adipose extracted from the M. semimembranosus muscle; 12 of 17 fatty acids investigated were affected (Karamichou et al. 2006). In a mapping population of cattle in Texas, Taylor et al. (1998) investigated the growth hormone gene GH1 on Chromosome 19 as a candidate gene for fat-related traits, including the proportion of unsaturated to saturated fat, and ether-extractable fat percentage, within subcutaneous fat over the l. dorsi muscle. The authors recommended that acetyl-CoA carboxylase and fatty acid synthase be tested as candidate genes for fat-related beef carcass traits. Further suggestions for fat-related traits in cattle came from Roy et al. (2005) in relation to fatty acid synthase on Chromosome 19 and mitochondrial glycerol-3-phosphate acyltransferase on Chromosome 26.
The aim of the present study was to investigate genetic differences in fatty acid composition using three resource populations of cattle. Phenotypes were recorded for fatty acid composition in the subcutaneous adipose tissue or milk fat from approximately 4800 cattle, and a screen with microsatellite markers on Chromosome 19 was conducted on a subset of these animals. This report describes the detection of a QTL in beef cattle affecting fatty acid composition of adipose fat, its validation in milk fat in independent populations of dairy cattle, and an investigation of fatty acid synthase as the causative gene for the QTL.
Methods
J × L backcross (Trial 1)
The trial design involved two Bos taurus breeds, Jersey (J) and Limousin (L), extreme for milk production and for muscling, respectively. A reciprocal backcross experiment was performed by mating six first-cross (F1) bulls (J × L or L × J) to both J and L cows. Three of the F1 sires were mated to dams in New Zealand, and the other three were mated to dams in Australia. Trial 1 comprises the New Zealand calves; most were generated by artificial insemination, but the 1996 ¾-Limousin calf crop was produced by embryo transfer using Hereford × Friesian cows as recipients, subsequently calving both single and twin calves. A total of 261 experimental backcross calves (162 ¾-Jersey and 99 ¾-Limousin) were born in Spring 1996, with another 155 born in Spring 1997 (102 ¾-Jersey and 53 ¾-Limousin). These 416 animals (heifers and steers) grazed on pasture and were supplemented with some roughage in winter (no concentrate feeding). Fatty acid percentages were determined (as described below) in adipose tissue sampled from subcutaneous fat collected post mortem in 1998 and 1999.
Milk composition trial (Trial 2)
A milk composition trial (Trial 2) was performed between November 2000 and February 2001, resulting in the collection of phenotypes during midlactation from approximately 4100 Friesian-sired cows from 21 commercial New Zealand dairy herds. In some cases separate grazing groups were used within a herd and the cow allocation to each of these groups was recorded. All cows were daughters of 11 Friesian sires that were used widely in New Zealand to supply semen for artificial insemination. The cows ranged in age from 2 to 11 years. One pooled milk sample (combining the evening and following morning milk) was collected from each cow in midlactation for the present study.
Milk composition trial (Trial 3)
Another milk composition trial (Trial 3) was performed in March 2003 using a single AgResearch herd at “Flock House,” Bulls, Manawatu Province, New Zealand, with a total of 248 cows’ milk sampled from an evening and the following morning milkings. The animals in this study ranged in age from 2 to 9 years and consisted of approximately equal numbers of Friesian-sired and Jersey-sired cows in late lactation. All cows grazed in one of two grazing groups (both breeds in both grazing groups) on a ryegrass/white clover diet.
Extraction and analysis of fatty acids
In Trial 1 (adipose fat), each fat sample was blended in a food processor and filtered through Whatman 541 filter paper (Whatman International Ltd, United Kingdom) in an oven set at 60°C to separate the fat. The fat layer was then transferred into tubes which were flushed with nitrogen, capped, and stored at −30°C for later analysis. In Trials 2 and 3 (milk samples), 1 ml of 0.15 g/ml EDTA was added to each 40 ml-sample of fresh milk, mixed, and refrigerated overnight. In the morning, samples were centrifuged at 1500 rpm for 4 min. The thick cream layer was transferred by spatula into 2-ml disposable microfuge centrifuge tubes and centrifuged twice at 15,000 rpm for 3 min, with the fat melted in an oven at 60°C between the centrifugations. The melted fat layer was then transferred into tubes that were flushed with nitrogen, capped, and stored at −30°C for later analysis.
Fatty acids were converted to their methyl esters, and individual fatty acid percentages in each sample were assayed with a gas chromatograph/flame ionization detector. Peak areas on the chromatographs were integrated to quantify the percentages of each fatty acid. The identity of each fatty acid was determined by comparing the retention time of each peak with known standards.
DNA extraction
In Trial 1, DNA was extracted from the sires and the two birth cohorts of calves using blood samples collected in their first year of life, using the method of Montgomery and Sise (1990). The exceptions were the ¾-Limousin calves born in 1996 from which DNA was extracted from ear cartilage using a Qiagen DNeasy Tissue Kit following the manufacturer’s instructions. This method was used because of the possibility of blood mixing in twin-born embryo-transfer calves. In Trial 2, DNA samples from 9 of the 11 Friesian sires were obtained from seminal fluid using a standard proteinase-k, phenol/chloroform extraction. DNA samples from all daughters were extracted directly from individual milk samples by standard methods or were isolated from milk blotted onto Whatman FTA paper according to the manufacturer’s instructions (Whatman FTA Technology, Whatman International Ltd, United Kingdom). In Trial 3, DNA was extracted from blood samples taken from the cows at the time of their milk sampling for this study, using the method of Montgomery and Sise (1990).
Genotyping of microsatellite markers
For Trial 1, all animals were genotyped for eight microsatellite markers: BM9202, HEL10, BMS2142, BP20, BM17132, BMS1069, ETH3, and BMS601 (Kappes et al. 1997). For Trial 2, the sires and a subset of the cows were genotyped for six microsatellite markers: BM9202, BMS2142, BM17132, BMS1069, ETH3, and BMS601.
Identification of single nucleotide polymorphisms (SNPs)
To identify SNP markers, a region of the fatty acid synthase (FASN) gene (GenBank accession No. AF285607) from one informative Friesian sire was resequenced. Primers (5′-GGTGCT CGAGGGTGTGGTGCA-3′ and 5′-TGCCGGAATT CCCGCATGGA-3′) were designed based on the FASN sequence and were used for PCR amplification. Each primer was designed so that the PCR product would have Xho1 and EcoR1 restriction endonuclease sites at alternate ends. The PCR fragments (approximately 2 kb in size) were amplified from genomic DNA in a 20-μl reaction, purified with a High Pure PCR Product Purification Kit (Roche), and digested with Xho1 and EcoR1 restriction endonucleases, and the enzymes were inactivated by heating for 20 min at 70°C. The sample was ligated into the plasmid pBluescript KS (Stratagene) and transformed into chemically competent E. coli strain XL1-Blue. Individual colonies were analyzed by colony PCR. Plasmid DNA was purified from E. coli, and the insert DNA from four individual colonies was sequenced bidirectionally from the T7 and T3 primer sites in the pBluescript plasmid. Typically, each sequencing reaction gave 800-900 bases of reliable sequence. Hence, in total we resequenced approximately 1.8 kb of the bovine FASN gene. One exception to this was SNP6, which was identified by direct sequencing of PCR product. The primers used in amplification were 5′-TGCTGTC CATGCGGGAAATC-3′ and 5′-AACTGCTGCAT GAAGAAGCAC-3′. The PCR product was purified with a PCR Product Purification Kit (Roche) and sequenced with a primer with the sequence 5′-GCTCACAATCCACAAGCTG-3′. All sequencing was done with an ABI 3100 DNA sequencer and products from Applied Biosystems (Foster City, CA). The sequences were analyzed by alignment using programs from the Wisconsin Package v10.2 [Genetics Computer Group (GCG), Madison, WI]. Table 1 lists the names given and the descriptions for the polymorphisms identified, consisting of SNPs and a dinucleotide insertion. Assays were designed to genotype these SNPs in the animals from Trials 1-3 via the Sequenom MASSARRAY system. The primers for these assays are also given in Table 1, along with the FASN SNP positions, so that their order was SNP4, SNP5, SNP3, SNP2, and SNP6. Haplotypes of sires were determined by using homozygous SNP data in progeny, with flanking markers adding more information.
Statistical analyses
In Trial 1, seven major fatty acids analyzed were myristic acid (C14:0), palmitic acid (C16:0), palmitoleic acid (C16:1), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), and linolenic acid (C18:3). In Trials 2 and 3, other fatty acids were also included, namely, those with 4-12 carbons, C4:0, C6:0, C8:0, C10:0, and C12:0. Analyses were performed first using SAS (1995) procedures to determine the important fixed effects in each trial.
For Trial 1, the fatty acid composition data were adjusted for slaughter group (incorporating year of birth and sex combinations) and breed of dam (in combination with herd of origin and birth rank), and adjusted values were calculated in residual standard deviation (RSD) units. After the FASN SNPs were genotyped, the combined microsatellite and SNP data were analyzed for linkage on a within-sire basis using QTL Express software (Seaton et al. 2002). The 95% and 99% thresholds were calculated by iterations (1000×), and bootstrapping was used to determine confidence intervals for QTL locations.
For Trial 2, effects were fitted for herd of origin (and associated grazing group), age of cow, Friesian proportion (accounting for crossbreds and grade-up animals, all with a Friesian sire), and a covariate for days-in-milk. Because of the batch processing of milk fat samples in preparation for gas chromatography, an adjustment for methylation batch was also required in the analysis. Adjusted values were generated in RSD units. After analyzing the six microsatellite markers, the FASN SNPs were genotyped over a subset of animals (consisting of those cows at the phenotypic extremes for milk fat percentage), and results were analyzed on a within-sire basis using QTL Express software.
A correlation analysis among residuals for 11 fatty acids was performed, followed by a principal components analysis to identify a possible first principal component that might be tested as another trait using QTL Express software.
For Trial 3, breed of cow, age of cow, and grazing group were used to generate standardized residuals; the covariate for date of calving was found to be nonsignificant. Statistical analyses are described below.
From each trial the SNP data alone (using RSDs) were then analyzed using chi-squared and least-squares procedures (regressing phenotype on genotype), to test the association between a SNP locus (or inherited haplotype from a parent) and a fatty acid component.
Results
Results from the three separate trials that identify and then validate a QTL for fatty acid composition are presented. Resource populations measured for fatty acid composition were those from Trial 1 (using subcutaneous adipose tissue) and Trials 2 and 3 (using milk fat).
Discovery of a QTL for fatty acid composition in adipose tissue (Trial 1)
Table 2 shows the overall means, RSDs, and breed-cross comparisons for fatty acid composition in subcutaneous adipose fat from Trial 1. All seven fatty acids analyzed, except myristic and palmitic acids, were affected significantly by breed-cross, with the ¾-Jersey cross having higher values of palmitoleic acid (C16:1) and oleic acid (C18:1), mainly compensated for by lower values of stearic acid (C18:0).
In Trial 1, preliminary analyses detected a QTL on bovine chromosome 19 that significantly affected one fatty acid (C14:0) at the 99% experiment-wise level, using microsatellites and linkage analysis (data not shown because more complete data are given in Table 3). Microsatellite BMS1069 was not informative for one of the three sire groups in Trial 1. Three other fatty acids were significant or approached significance at the 95% experiment-wise level (C16:1, C18:0, and C18:1). All QTL that were detected were located at approximately 70 cM.
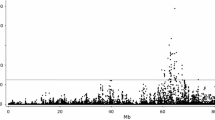
The FASN gene was identified as a positional candidate gene for this QTL because the location of FASN on bovine Chromosome 19q22 (Roy et al. 2001, 2005) coincided with the estimated location of the observed QTL, and because variation in FASN activity might explain the animal-to-animal variation in fatty acid composition. The cattle FASN gene is in approximately the same location as the microsatellite marker IOBT34 (at ∼77 cM on the map of Ihara et al. 2004), with marker BMS1069 about 1-2 cM from IOBT34 (Roy et al. 2005). Six SNPs in the cattle FASN gene were identified by resequencing and five of these were found subsequently to be polymorphic in our populations (Table 1). The probability values for four fatty acids are shown on a log10 scale in Fig. 1, from the combined analyses of microsatellites and SNPs on Chromosome 19 by position (substituting a FASN SNP for microsatellite BMS1069), and the within-sire group results are summarized in Table 3. The QTL effect on C14:0 accounted for a difference of about 0.38% ± 0.07% of all adipose fat (0.85 RSDs or 11.3% of the C14:0 mean), and the squared correlation coefficient indicated that the QTL accounted for 17% of the total variation in adjusted values for C14:0.

Log10-transformed probabilities for fatty acid composition traits in adipose fat across Chromosome 19 within sire groups (Trial 1). Fatty acids were (a) residuals for C14:0 for sire group 402 (—); (b) residuals for C18:1 for sire group 394 (- - - ); (c) residuals for C16:1 for sire group 402 (· · · ·); (d) residuals for C18:0 for sire group 417 (+ + +).
Validation of the QTL in milk (Trial 2)
The predominant fatty acids in the milk fat samples of Trial 2 were C16:0 30.5% (RSD = 2.7%), C18:1 15.7% (1.8%), C14:0 12.6% (0.8%), and C18:0 9.5% (1.3%). The most significant linkage results using both microsatellites and SNPs combined are given in Table 4 (six of these fatty acids were significant at p < 0.01 and four at p < 0.05). The probability values for the six most significant fatty acids for one Friesian sire, Sire A, are shown on a log10 scale in Fig. 2, from the combined analyses of microsatellites and SNPs on Chromosome 19 by position; microsatellites BMS1069 and ETH3 were not informative for this sire. A QTL was identified that affected C10:0, C12:0, C8:0, C14:0, C18:2, and C18:1 (in order of significance), within the progeny of Sire A. The estimated location of this milk fat QTL coincided with the QTL observed for adipose fat in Trial 1.

Log10-transformed probabilities for fatty acid composition traits in milk fat across Chromosome 19 for Friesian Sire A (Trial 2).
In Trial 2, a correlation analysis among residuals was performed for the ten fatty acids that had a significant QTL. The largest positive correlations were as follows: C6:0 × C8:0, 0.70; C8:0 × C10:0, 0.83; C10:0 × C12:0, 0.96; C12:0 × C14:0, 0.76; C8:0 × C12:0, 0.70; C10:0 × C14:0, 0.74; and C18:1 trans × CLA, 0.78. The two most negative correlations were between C12:0 and C18:1 (−0.54), and C14:0 and C18:1 (−0.51). A principal components analysis showed that the first principal component accounted for 37% of the variance among the fatty acids, the second a further 18%, and the third a further 15%. The first principal component was used as a variate to plot a new QTL map and this gave a very similar plot to that for C10:0 in position and shape in Fig. 2. The maximum height was slightly greater for this variate than for C10:0 (p = 2.53E-09 and p = 7.92E-09, respectively, i.e., a smaller probability for the first principal component).
Linkage disequilibrium analysis
Trial 1
Analysis of SNPs in Trial 1 animals led to the identification of a series of five-SNP haplotypes. The three sires in Trial 1 had a combined total of three haplotypes for SNP2-SNP6 (Table 5). Using SNP and informative microsatellite flanking markers together, the numbers of animals with each haplotype are also given by sire in Table 5. Each sire carried the haplotype CGCDA (ordered by nucleotide sequence from their positions in Table 1 as SNP4, SNP5, SNP3, SNP2, and SNP6, respectively; D = deletion) from the Limousin parent, and the paired chromosome from the Jersey parent carried one of two haplotypes, either the CGTIG or the AATIG nucleotide sequence (I = insertion). Numbers of progeny receiving the two haplotypes from sire 394 appeared to be imbalanced because only the third, fourth, and fifth SNPs (i.e., SNP3, SNP2, and SNP6, respectively) of the haplotype were informative. Also, the high level of heterozygosity from the sire-394 haplotype CGCDA, when paired with chromosomes from Jersey cows carrying mainly the CGTIG or the AATIG haplotype, meant that the haplotype inherited from the sire could not be derived. The haplotype inherited from the dam of each backcross calf in Trial 1 was determined by subtraction. There was a predominance of CGTIG haplotypes among dams, with 138 progeny (144 when including information from flanking markers), followed by the AATIG haplotype with 35 (50) progeny, and then the CGCDA haplotype with 15 (39) progeny.
The haplotype difference in C14:0 fatty acid percentage in subcutaneous adipose fat (Table 6, top panel) was highly significant (p < 10−6) for sire 402, and it approached significance in the same direction for sire 417 with the same haplotype comparison (p = 0.07). The haplotype effect, CGCDA minus CGTIG, was also significant for sire 394 (p = 0.02), but it was in the opposite direction from the contrast of CGCDA minus AATIG. Sire 394 was homozygous for SNP4 and SNP5, so the contrast examined may effectively have represented another haplotype.
Table 6 also shows results from sire and dam haplotype effects fitted together [second panel, with fewer animals than for sire effects (top panel) because there were some animals with unknown dam haplotype], along with the sire and dam SNP4 effects fitted together (bottom panel). Results for SNP4 and SNP5 were identical because of their very close linkage. The size of the effect of nucleotide substitution C minus A at SNP4 for sire 402 was 0.75 ± 0.18 RSD, or 0.34% ± 0.08% of all fat (i.e., 9.9% of the mean for C14:0 in adipose fat). The size of the effect for sire 417 was about half of that for sire 402, and in the same direction. The sign of the C-minus-A substitution in independent dams was the same as in the sires, and it was significant among the Jersey dams (p = 0.008).
Trial 2
The same five SNPs and the haplotypes for FASN were tested for an association with milk fat phenotype in Trial 2 cows. Table 7 (top panel) shows the results for association of all cows genotyped, combining daughters of Sire A and other available Friesian sires. SNP4 and SNP5 effects were both significant (p < 0.05), with effects on C14:0 percentage of −0.28 ± 0.14 RSD units (for CC minus AA) or −0.33 ± 0.14 RSD units (for GG minus AA), respectively. In the largest Friesian sire group evaluated in Trial 2 (sire A, carrying haplotypes CGCDA and AATIG), 111 daughters inherited the CGCDA haplotype from the sire, and 107 daughters inherited the AATIG haplotype. The difference in C14:0 percentage in milk fat between the haplotypes CGCDA and AATIG was significant (p < 0.001), with an effect of −0.42 ± 0.13 RSD units (Table 7, bottom panel). Fatty acids C6:0, C8:0, C10:0, C12:0, and C18:1 were also affected by these sire haplotypes at similar or more significant probabilities to C14:0 (Table 7) and with similar sizes of genotypic effects (on an RSD basis). As was found in Trial 1, the effect on C14:0 was in the opposite direction of the effect on C18:1.
Trial 3
The FASN SNPs were further validated for the milk fat phenotype by genotyping the Trial 3 cows. The predominant fatty acids found in milk fat from Friesian-sired and Jersey-sired cows in this trial were C16:0 29.3% (RSD = 2.1%), C18:1 19.1% (2.3%), C14:0 10.1% (0.9%), and C18:0 10.1% (1.4%). Overall, 94% of the 121 Friesian-sired cows and 68% of the 113 Jersey-sired cows whose DNA was analyzed were purebred or ¾ bred. Table 8 shows the genotype numbers and allele frequencies for the five SNPs in each breed.
Over both breeds combined (data not tabulated), SNP3 was associated with C18:0 percentage (p < 0.05), while SNP4 and SNP5 were both significant for percentages of C14:0 and of C18:1 (all p < 0.05). When analyzing within breed (Table 9), only the Jersey data were significant, as follows: the genotype effect (CC minus AC) on C14:0 percentage in milk fat was −0.64 ± 0.19 RSD units (p = 0.0009) for SNP4, and the genotype effect (GG minus AG) on the same trait was −0.70 ± 0.20 RSD units (p = 0.0007) for SNP5. SNP4 and SNP5 in Jerseys had opposite effects on C18:1 compared with their effects on C14:0, i.e., respectively, 0.56 ± 0.19 RSD units (p = 0.0046) and 0.62 ± 0.20 RSD units (p = 0.0027) for C18:1. Consequently, an allelic substitution at SNP4 (A instead of C) or SNP5 (A instead of G) led to an increase in the C14:0 percentage in milk fat and a corresponding reduction in the C18:1 percentage in milk (RSD units). Converting RSD units back to the units of measurement, a SNP4 or SNP5 substitution led to a 2.8-fold greater change in C18:1 concentration than in C14:0 concentration. The same trends appeared to apply to Friesians as to Jerseys, although contrasts were not significant in Friesians.
Discussion
The aim of this study was to identify QTL that explain some of the genetic variation in fatty acid composition in cattle. Initially, we conducted a screen of Chromosome 19 using a reciprocal Jersey × Limousin backcross (Trial 1), with phenotypes for subcutaneous adipose fatty acid composition. A significant QTL was identified for two fatty acids (Table 3) on Chromosome 19, and a candidate gene for this QTL was identified as FASN, a central multifunctional enzyme consisting of seven distinct catalytic sites and responsible for de novo fatty acid biosynthesis (Roy et al. 2001). Genetic variants of FASN have been reported recently to be associated with fat percentage in bovine milk (Roy et al. 2006). Using different resource populations, the present study shows that FASN, or possibly a closely linked gene, affects adipose fat composition and milk fat composition. The significant QTL effects in milk fat were also found to extend to short-chain fatty acids (C6:0, C8:0, C10:0, and C12:0), which do not exist in adipose fat.
Having identified a QTL on Chromosome 19 by using microsatellites and having posited a candidate gene, a haplotype from a set of five SNPs from the FASN gene was then tested for both linkage and association, and the haplotype results are shown in Table 6 for Trial 1. Significant results are shown for both sire and dam effects. Two SNPs in particular, SNP4 and SNP5, showed large effects (up to 0.8 RSD units, with the power of the design sufficient to reach statistical significance with an effect of about 0.4 RSD or more).
It should be noted that of the SNPs discovered in the FASN gene herein, only SNP6 encodes an amino acid change (Table 1). As the other SNPs are unlikely to affect the enzyme activity of the fatty acid synthase protein or the expression of the FASN gene, the most likely scenario is that SNP4 and SNP5 are in strong linkage disequilibrium with the true functional variant. However, there are no other obvious candidate genes in the immediate vicinity or other potentially causative SNPs in the region.
From the within-sire analyses (Trial 1, Table 3; and Trial 2, Table 4), we showed that the confidence intervals were very wide, often up to 50 cM. Accordingly, from the sire families alone, there was no way of distinguishing between the genes FASN and GH1. However, from the SNP data, the likely interval for strong linkage disequilibrium is acknowledged as less than 0.3–0.5 cM for cattle breeds such as the Holstein-Friesian and Jersey (Goddard et al. 2006; Spelman and Coppieters 2006). Because the mapped distance between FASN and GH1 is 3.8 cM, it would be unlikely that FASN SNPs are in significant linkage disequilibrium if the real gene was GH1. In addition, we analyzed FASN SNP4 data from dams in Trial 1 (Table 6), and the effect from Jersey dams was also significant. Strong linkage disequilibrium is also unlikely to be observed consistently across breeds (Trials) at this distance.
The same five SNPs were also tested in two cattle resource populations with milk fat phenotypes to validate the linkage and association with the SNPs. SNP4 and SNP5 were again found to show associations with C14:0 percentage in milk fat. They were consistent in the direction of the effects in milk fat for both SNP4 and SNP5 across Trial 2 (Table 7; a significant effect in Friesians) and Trial 3 (Table 9; a significant effect in Jerseys, with Friesians in Trial 3 carrying the same sign of effect, although not significant). However, an important finding was that the SNP allelic effects on C14:0 percentage in the milk fat of Trials 2 and 3 (Tables 7 and 9) were opposite in sign from those found for C14:0 percentage in adipose fat in Trial 1 (Table 6). The “A” alleles of both SNP4 and SNP5 were associated with increased percentages of C14:0 in milk fat, but they had the opposite effect in adipose fat. This was probably a tissue expression effect, not a breed effect, because the Jersey breed was common to Trials 1 and 3.
The FASN protein in adipose tissue synthesizes mainly C14:0 and C16:0, whereas FASN in mammary tissue synthesizes fatty acids of chain length varying from C4:0 to C16:0 in 2-carbon increments. The thioesterase enzymes responsible for cleaving off the acyl groups from the FASN complex are different in bovine mammary FASN and adipose FASN (Smith 1994), although this is not well characterized. A possible mechanism for FASN is that SNP4 and/or SNP5 may be associated with thioesterase specificity for fatty acids, and this association might also be tissue specific. The level of regulation of FASN by external factors is different in adipose and mammary tissue, and this difference may be responsible for the different SNP effects on C14:0 in the two tissue types. For example, the FASN gene activity in situ is negatively regulated by C18:1 and other long-chain fatty acids (Hansen and Knudsen 1987; Gaynor et al. 1995), and C18:1 percentages are much higher (by a factor of ∼2.5) in adipose tissue than in milk fat.
Microsatellites on Chromosome 19 were significantly associated with C14:0 concentrations but not with C16:0 concentrations, in both adipose and milk fat. The association was also observed for C6:0, C8:0, C10:0, and C12:0 in milk. One explanation for this might be that the effect observed for C16:0 is confounded by contributions from dietary fatty acids, whereas dietary input for C6:0, C8:0, C10:0, C12:0, and C14:0 is less.
Conclusions
A QTL was identified for a series of fatty acids in adipose fat and milk fat on bovine Chromosome 19, and FASN was postulated as a candidate gene for the QTL. Five SNPs on FASN were significantly associated with fatty acid percentages, up to 0.8 RSD in magnitude, and the SNP4 or SNP5 substitution effects for C14:0 percentage were found to have an effect in the opposite direction in adipose fat than that of milk fat.
References
Baumgard LH, Kay JK, Moore CE, Roche JR, Vanbaale MJ, et al. (2005) Regulating milk fat synthesis: potential on-farm applications of basic science. Proc NZ Soc Anim Prod 65, 33–39
Berner LA (1993) Defining the role of milkfat in balanced diets. Adv Food Nutr Res 37, 131-257
Clop A, Ovilo C, Perez-Enciso M, Cercos A, Tomas A, et al. (2003) Detection of QTL affecting fatty acid composition in the pig. Mamm Genome 14, 650-656
Davies HM (1996) Engineering new oilseed crops from rapeseed. In Progress in new crops. Proceedings of the Third National Symposium, Indianapolis, IN, 22–25 October 1996, Janick J, (ed). (Alexandria, VA: American Society for Horticultural Science Press), pp 299–306
Gaynor PJ, Waldo DR, Capuco AV, Erdman RA, Douglass LW, et al. (1995) Milk fat depression, the glucogenic theory, and trans-18:1 fatty acids. J Dairy Sci 78, 2008–2015
Goddard ME, Hayes B, McPartlan H, Chamberlain AJ (2006) Can the same genetic markers be used in multiple breeds? In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production. Belo Horizonte, Brazil, August 2006 (CD-ROM summary)
Hansen H, Knudsen J (1987) Effect of exogenous long-chain fatty acids on individual fatty acid synthesis by dispersed ruminant mammary gland cells. J Dairy Sci 70, 1350–1354
Ihara N, Takasuga A, Mizoshita K, Takeda H, Sugimoto M, et al. (2004) A comprehensive genetic map of the cattle genome based on 3802 microsatellites. Genome Res 14, 1987-1998
Kappes SM, Keele JW, Stone RT, McGraw RA, Sonstegard TS, et al. (1997) A second-generation linkage map of the bovine genome. Genome Res 7, 235-249
Karamichou E, Richardson RI, Nute GR, Bishop SC (2006) Identification of quantitative trait loci affecting fatty acid composition in sheep. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production. Belo Horizonte, Brazil, August 2006, Conference Paper No. 13–12
Lee C, Chung Y, Kim J-H (2003) Quantitative trait loci mapping for fatty acid contents in the backfat on porcine chromosomes 1, 13, and 18. Molecules Cells 15, 62-67
Montgomery GW, Sise JA (1990) Extraction of DNA from sheep white blood cells. NZ J Agric Res 33, 437-441
Mozaffarian D, Ascherio A, Hu FB, Stampfer MJ, Willett WC, et al. (2005) Interplay between different polyunsaturated fatty acids and risk of coronary heart disease in men. Circulation 111, 157-164
Perez-Enciso M, Clop A, Noguera JL, Ovilo C, Coll A, et al. (2000) A QTL on pig chromosome 4 affects fatty acid metabolism: evidence from an Iberian by Landrace intercross. J Anim Sci 78, 2525-2531
Roy R, Gautier M, Hayes H, Laurent P, Osta R, et al. (2001) Assignment of the fatty acid synthase (FASN) gene to bovine chromosome 19 (19q22) by in situ hybridization and confirmation by somatic cell hybrid mapping. Cytogenet Cell Genet 93, 141-142
Roy R, Zaragoza P, Rodellar C, Gautier M, Eggen A (2005) Radiation hybrid and genetic linkage mapping of two genes related to fat metabolism in cattle: fatty acid synthase (FASN) and glycerol-3-phosphate acyltransferase mitochondrial (GPAM). Anim Biotechnol 16(1), 1-9
Roy R, Ordovas L, Zaragoza P, Romero A, Moreno C, et al. (2006) Association of polymorphisms in the bovine FASN gene with milk-fat content. Anim Genet 37, 215-218
SAS (1995) JMP v3 (Cary, NC: SAS Institute Inc.)
Seaton G, Haley CS, Knott SA, Kearsey M, Visscher PM (2002) QTL Express: mapping quantitative trait loci in simple and complex pedigrees. Bioinformatics 18, 339-340
Singh SP, Zhou XR, Liu Q, Stymne S, Green AG (2005) Metabolic engineering of new fatty acids in plants. Curr Opin Plant Biol 8, 197-203
Smith S (1994) The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB J 8, 1248-1259
Spelman R, Coppieters W (2006) Linkage disequilibrium in the New Zealand Jersey population. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production. Belo Horizonte, Brazil, August 2006 (CD-ROM summary)
Taylor JF, Coutinho LL, Herring KL, Gallagher DS JR, Brenneman RA, et al. (1998) Candidate gene analysis of GH1 for effects on growth and carcass composition of cattle. Anim Genet 29, 194-201
Temme EH, Mensink RP, Hornstra G (1997) Effects of medium chain fatty acids (MCFA), myristic acid, and oleic acid on serum lipoproteins in healthy subjects. J Lipid Res 38, 1746-1754
Acknowledgments
The authors thank the Farm Managers and their staff at AgResearch’s Tokanui Station (Trial 1) and Flock House Station (Trial 3) for care of the animals and for their assistance with data and sample collection. They thank AgResearch’s staff at the Dairy Science and MIRINZ Centres at Ruakura for carrying out the fatty acid composition analyses, and also thank Dr. Paul Shorten for advice during manuscript preparation. This work was funded by the New Zealand Foundation for Research, Science & Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Morris, C.A., Cullen, N.G., Glass, B.C. et al. Fatty acid synthase effects on bovine adipose fat and milk fat. Mamm Genome 18, 64–74 (2007). https://doi.org/10.1007/s00335-006-0102-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-006-0102-y