
Abstract
Body mass (BM) is a classic polygenic trait that has been extensively investigated to determine the underlying genetic architecture. Many previous studies looking at the genetic basis of variation in BM in murine animal models by quantitative trait loci (QTL) mapping have used crosses between two inbred lines. As a consequence it has not been possible to explore imprinting effects which have been shown to play an important role in the genetic basis of early growth with persistent effects throughout the growth curve. Here we use partially inbred mouse lines to identify QTL for mature BM by applying both Mendelian and Imprinting models. The analysis of an F2 population (n ≈ 500) identified a number of QTL at 14, 16, and 18 weeks explaining in total 31.5%, 34.4%, and 30.5% of total phenotypic variation, respectively. On Chromosome 8 a QTL of large effect (14% of the total phenotypic variance at 14 weeks) was found to be explained by paternal imprinting. Although Chromosome 8 has not been previously associated with imprinting effects, features of candidate genes within the QTL confidence interval (CpG islands and direct clustered repeats) support the hypothesis that Insulin receptor substrate 2 may be associated with imprinting, but as yet is unidentified as being so.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Body mass (BM) is an important phenotypic trait because it has a pervasive effect on almost all other aspects of animal physiology and ecology. As a classic polygenic trait, there has been sustained interest in understanding the genetic basis of its variation for over a century (since Galton 1889), probably initially stimulated by the fact that the trait is easily measured, and more recently because it was recognized that the normal distribution of the phenotype fits the quantitative genetic model for continuous traits reasonably well (Falconer and Mackay 1996). However, our understanding of the genetic basis of variation in BM is still far from being complete. Studies of the genetic basis of BM have indicated that different genes are involved in different phases of growth (Cheverud et al. 1996; Vaughn et al. 1999; Rocha et al. 2004) and there appears to be some negative interaction between early (0-20 days) and late (20-70 days) acting genes during these two growth phases (Cheverud et al. 1983; Leamy and Cheverud 1984). Similar differential genetic age dependencies have been found in humans younger and older than 20 years (Rice et al. 1999). Relatively few studies, however, have addressed the genetic factors that influence BM of mice during the period beyond 70 days of age. This is unfortunate because it is during this phase that the gradual accretion of body fatness occurs (Bunger et al. 1998b), a situation that is paralleled in the human growth curve. Therefore, understanding the genetic factors that influence body mass at this age is of considerable importance for our understanding of the genetic component contributing to the global obesity epidemic.
Monogenic animal models, including natural loss-of-function mutations and artificially produced transgenic overexpressing and knockout mice, have been an important resource in elucidating the physiology underpinning phenotypic variation in BM (Doolittle et al. 1996; Efstratiadis 1998; Snyder et al. 2004). However, there are limitations in drawing conclusions about normal functional effects from these major genetic perturbations because these genes rarely act alone but interact with other genes in complex physiological pathways (Efstratiadis 1998). In terms of determining areas of the genome associated with population variation in BM and body composition, genetic analysis of crossbred populations of mice, rat, and pig (for examples see Snyder et al. 2004) has led to the identification of a number quantitative trait loci (QTL). In human populations, QTL for BM (and in particular BM index) have been identified (Snyder et al. 2004), some of which correspond to model animal species QTL by synteny [e.g., the BMI QTL on human 4p12 (Deng et al. 2002) corresponds to Chromosome 5 mouse BM QTL BW8 (Brockmann et al. 1998) and Bwem1, a QTL for male 30-day BM (Le Roy et al. 1999)].
In model animal species, the basic starting point for many QTL mapping studies is to take two inbred lines that differ for the phenotype of interest and produce a segregating population (including F2, backcross and recombinant inbreds). The F2 intercross has proven to be the most frequently used population design because it allows both additive and dominance QTL effects to be estimated and it allows exploration of pleiotropy (Brockmann et al. 2000; Leamy et al. 2002) and epistatic interactions (Brockmann et al. 2000; Yang 2004; Ishikawa and Namikawa 2004). However, using an F2 design between two inbred lines precludes the study of imprinting effects, where parental-specific epigenetic modifications of DNA elements control the differential expression of maternal and paternal alleles because it is impossible to determine the parent of origin of any allele. As a consequence, the effect of imprinting has not been explored significantly in rodent models, despite substantial estimation in F2 pig population studies (Jeon et al. 1999; Rattink et al. 2000; Quintanilla et al. 2002; Thomsen et al. 2004). The potential importance of imprinting effects in QTL expression was demonstrated by De Koning et al. (2000) using an F2 population generated by crossing two outbred and genetically divergent pig breeds; four of the five body composition QTL identified were associated with significant imprinting effects.
In this study, partially inbred mouse lines resulting from a long-term selection experiment were used in a genome-wide QTL mapping analysis including estimation of imprinting effects. The parental lines were divergently selected for food intake between 8 and 10 weeks corrected for mean BM (Hastings et al. 1997; Bunger et al. 1998a). At generation 47, Selman et al. (2001) showed both male (M) and female (F) high-intake (H) mice at approximately 18 weeks were significantly heavier (HM = 33.5 ± 0.46 g; HF = 34.2 ± 0.89 g) than low-intake (L) contemporaries (LM = 31.0 ± 0.62 g; LF = 31.2 ± 0.62 g). Although this line divergence is relatively small (6% and 9% of mean BM in males and females, respectively), QTL influencing the trait may diverge in the two lines as a result of pleiotropic effects and linkage. As a consequence both positive and negative effects within the high and low line may be present resulting in only a minor net BM divergence. The selection for food intake corrected for BM in mature animals has also resulted in a significant pleiotropic response in resting metabolic rate (RMR), where both male and female high-intake mice were also associated with higher RMR (Bunger et al. 1998a; Selman et al. 2001). As a result we postulate that BM-associated QTL identified from a cross of these selection lines may play a more important role in energy balance than those identified in segregating populations made between lines where BM is highly divergent.
Materials and methods
Mouse lines and phenotypic measurements
The mouse lines used in this study (the Maintenance-lines or M-lines) have been divergently selected for high (MH) and low (ML) food intake between 8 and 10 weeks of age, corrected by phenotypic regression on mean BM (Hastings et al. 1997). The base population was created from a cross of the six control lines (random mating for 20 generations) for the Edinburgh divergent selection lines. The Edinburgh selection lines, including the control lines, were formed from an F1 between two inbred lines (JU and CBA) crossed with an outbred population (CFLP) (Sharp et al. 1984). For the first 24 generations of selection, the M-lines were maintained as three replicates in each direction of selection. At this point the three replicates were intercrossed and only a single line in each direction was maintained. At generation 38 phenotypic selection was suspended and from generation 43 onward the lines were maintained by full-sib mating to produce inbred lines (MHi and MLi).
QTL mapping analysis was performed using an F2 intercross design. To ensure mice were the same age during measurements, and because of the time-consuming nature of subsequent measurements taken on the F2 population, the large sample size was generated from matings carried out in six different batch time points. Using the inbred lines developed and maintained at the University of Edinburgh, F1 crosses between MHi and MLi generations 54-60 (inbred generations 11-17 when the average inbreeding coefficient had reached 0.908-0.974), with the exception of generation 56, were made representing both reciprocal halves (MHi × MLi and MLi × MHi). After weaning (3 weeks of age), F1 individuals were transported to the rodent facility at the University of Aberdeen, where single-pair F1 matings were made at 12 weeks of age and included all the possible reciprocal half pairings. The F2 mice were maintained in their family groups until they were 3 weeks old at which time they were weaned into single-gender groups (nmax = 10) and identified using unique ear clips. Single-gender groups were kept in MB1 cages (45 cm × 28 cm × 13 cm; North Kent Plastics, Rochester, UK) under a photoperiod of 12 h:12 h light:dark (lights on at 07:00 h) at 20 ± 2°C. All individuals had access to water and food ad libitum (breeder and grower diet, Special Diets Services, BP Nutrition, Witham, UK). At approximately 14 weeks of age animals were individually housed in M3 cages (48 cm × 15 cm × 13 cm; North Kent Plastics) until the end of the experimental period. Sawdust bedding was provided at all stages. A total of 515 F2 individuals were produced from 51 F1 pairings. BM was recorded at three different time points, representing approximately 14, 16, and 18 weeks of age (there was minor variation in age around these time-points). Here we present data on the results of QTL mapping analysis for BM at 14, 16, and 18 weeks of age.
Genotyping
Genomic DNA was extracted from spleen tissue using DNAeasy Tissue Kit (Qiagen, Crawley, UK). All animals were genotyped using microsatellite MapPairTM primers purchased from ResGen (Huntsville, AL). DNA was amplified using REDTaq ReadyMix (Sigma-Aldrich, Gillingham, UK) and standardized PCR protocols: 40 ng of template DNA in a total reaction volume of 10 μl. The PCR amplifications were carried out using 96-well microtiter plates on a GeneAmp PCR-System 9700 (Applied Biosystems, Foster City, CA). The main amplification protocol was as follows: initial denaturization 96°C for 2 min, then 30 cycles at 94°C for 45 sec, 57°C for 45 sec, 72°C for 60 sec, ending with a final extension period at 72°C for 7 min. Products were run on 20-cm-long, vertical running 6% polyacrylamide gels (Protean II Xi 20 cm cells, Bio-Rad, Hemel Hempstead, UK) for 2-3 h at 240 V. Gels were photographed after 20 min of ethidium bromide staining using a Syngene Genegenuis Gel documentation system (Syngene, Cambridge, UK) under ultraviolet light. All gels were scored on two occasions and, where discrepancies were present, genotyping was repeated.
High- and low-line parents were used to identify markers segregating within the population. A total of 300 mouse MapPairTM primers were investigated yielding 87 informative loci where alleles segregate in more than 50% of families (lower levels of information content were tolerated in areas of poor map coverage). The average information content of markers was calculated according to Knott et al. (1998) using the QTL Express software (Seaton et al. 2002). Markers were chosen to be distributed over all chromosomes, yielding an average map distance of 20.5 cM (MGD 2004). Because the M-lines were not completely inbred and markers were still segregating within line, all individuals in the population were genotyped so that QTL mapping methodologies for outbred populations could be applied. For linkage analysis, population-specific marker maps were generated using CriMap 2.4 (Lander and Green 1987).
Data analyses
BM at 14, 16, and 18 weeks (14wk, 16wk, 18wk) was analyzed to identify the magnitude and location of QTL. All analysis was carried out using GenStat 7.1 (GenStat 7 Committee 2003). Data were investigated for normality and transformed by box-cox transformation where appropriate. The most appropriate model for analysis was identified using the Wald statistic in the residual maximum likelihood (REML) directive. The effects investigated included the fixed effects associated with gender (levels = 2), number in litter at weaning (levels = 12), reciprocal half of mating structure (HL × HL, HL × LH, LH × LH or LH × HL; levels =4), and batch effect (representing the six different mating time points), and the random effects of dam (n = 51) and the covariate of age in days (to account for variation in age at weighing). The effects of gender and litter size, random effect of dam, and covariate of age were found to be significant for all BM measurements and were included in the GenStat 7.1 (GenStat 7 Committee 2003) linkage analysis model.
Data were analyzed using a methodology for analysis of crosses between outbred lines as described by Haley et al. (1994) where progenitor lines are assumed homozygous for different QTL alleles but markers may segregate within lines. Initial analysis fitted single Mendelian QTL in a standard interval mapping model. The F-ratio test statistic was used to determine QTL position at 1-cM intervals. Although Wald tests may be more appropriate for REML analysis, correlations between Wald tests and F-ratios were high (r2 > 0.99), thus, F-ratios were used as an established QTL mapping test statistic. The F-ratio was generated using a reduced model (including fixed effects of gender and litter, random effect of dam, and covariate of age) versus a full model where all effects fitted in the reduced model were included plus coefficients representing additive (Pa) and dominance (Pd) components, resulting in 2 degrees of freedom (df) in the numerator. Following the standard single-QTL analysis, estimates were revised by fitting background genetic effects to remove variance due to QTL elsewhere on the genome (Jansen 1993; Zeng 1994). These background genetic effects were included as cofactors in the analysis, beginning with the locus showing the largest estimated variance followed by the second largest effect, until no further loci were identified surpassing the suggestive threshold. Following this process, a cofactor was removed from analysis when estimating its own QTL position and effect. Interaction terms were tested for gender by additive genetic and dominance effects.
Mapped QTL were also tested for the effects associated with imprinting, where the QTL effect is dependent on the parental line of origin. Models testing the effects of maternal and paternal imprinting were explored according to De Koning et al. (2000). In brief, to separate the probability of an individual inheriting an allele from its sire or dam, saturated Imprinting models including paternal (Ppat), maternal (Pmat), and dominance (Pd) components were fitted. The presence of imprinting was tested initially using the F-ratio of imprinting effects versus no QTL present on the chromosome, yielding 3 df in the numerator of the F-ratio. Where imprinting effects were indicated, the Imprinting model was compared with the single Mendelian QTL model to test the significance of imprinting effect, yielding an F-ratio with 1 df in the numerator. To explore the individual contribution of maternal and paternal imprinting, maternal (Pmat + Pd) and paternal (Ppat + Pd) specific Imprinting models were also applied (De Koning et al. 2002), which were associated with 2 df in the numerator. Finally, models testing the presence of two QTL on a chromosome were applied as proposed by Haley and Knott (1992) using an F-ratio testing the two-QTL model against the one-QTL model. The two-QTL models were applied to only QTL reaching the significant (p < 0.05) threshold and scans were carried out at 2-cM density.
Genome-wide empirical thresholds were estimated for significant (p < 0.05) and highly significant (p < 0.01) using the permutation test proposed by Churchill and Dorge (1994). To apply consistent thresholds to all traits, marker data were permutated (n = 5000) against a simulated normally distributed phenotype with mean of 0 and variance of 1. In addition to the genome-wide significance threshold, a threshold representing suggestive linkage was determined as proposed by Lander and Kruglyak (1995). The suggestive level represents the probability of one false-positive result per genome-wide scan. In a genome of 20 chromosomes pairs, this is approximately equivalent to a chromosome-wide significance threshold of p < 0.05. Significance thresholds for the Imprinting models were calculated as described in Quintanilla et al. (2002). Confidence intervals for QTL position were determined using the 1-LOD dropoff method. This method has been shown to be approximate to a 90%-95% confidence interval (van Ooijen 1992; Visscher et al. 1996).
Results
Marker information
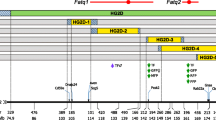
A total of 300 markers were investigated to identify 87 markers segregating between lines (79 loci with 2 alleles and 8 loci with 3 alleles). Complete marker information was available for all parental MHi and MLi and F1 individuals, and the average map coverage for the F2 population was 97%. The average information content of the markers genotyped on the F2 population was found to be 0.79 for additive and 0.72 for dominance effects. Estimation of imprinting effects was associated with lower information content being zero at 43% of markers (with no information for Chrs 18 and 19) and 0.24 for the remaining marker loci. The population-specific map distances estimated using CriMap (Lander and Green 1987) resulted in an expanded total map distance and increased mean marker density of 23.6 cM (Fig. 1). However, there were only two large changes in map distance with the distance betweeen D9MIT129 and D9MIT136 increasing from 26 to 42.3 cM and the distance between D2MIT106 and D2MIT266 increasing from 33.4to 56.1cM and resulting in the largest marker interval.

The map of all markers genotyped in the pedigree. Marker positions are given according to those calculated from the pedigree specific data using CriMap (Lander and Green 1987).
Phenotypic variables
Phenotypic information for the traits analyzed was available on between 435 and 515 of the F2 population (Table 1). All data were transformed to normality using box-cox transformation to remove small significant deviations from normality. No biological outliers were identified within the data set. All BM traits showed moderate variability with coefficients of variation (CV) of approximately 16%. The phenotypic correlations between the different measures of BM were high, being 0.988 between 16wk and 14wk, 0.914 between 18wk and 14wk, and 0.906 between 16wk and 18wk.
The location of mapped QTL
The empirical genome-wide significance thresholds generated by permutation were 7.56 for significant (p <0.05), 9.41 for highly significant (p < 0.01), and a suggestive threshold (Lander and Kruglyak 1995) of 4.32. Using the suggestive linkage threshold, a total of 23 QTL were identified on 11 of the 19 autosomes; no X-linked QTL were identified (Table 2). Of these, 9 QTL were significant (p < 0.05) and 5 surpassed the highly significant threshold (p < 0.01). The total phenotypic variance explained by the mapped QTL was 31.5%, 34.4%, and 30.5% for BM at 14, 16, and 18 weeks, respectively.
For all three BM traits QTL have been identified on Chrs 5, 7, 8, 9, and 18 (Table 2). The QTL on Chr 5 at the most proximal end (73 cM) was highly significant for 16wk, explaining 4.89% of total phenotypic variance. On Chr 7 the QTL was associated with an increase in variance explained with the increase in age, from 3.6% at 14wk, 4.4% at 16wk, and the highly significant QTL at 18wk being associated with 4.7% of phenotypic variance. A QTL of large effect was identified on Chr 8 reaching maximum F-ratios of between 25.57 and 34.28 and explaining 12.34%, 12.29%, and 10.65% of total phenotypic variation for BM at 14, 16, and 18 weeks, respectively (Table 2; Fig. 2a). The QTL on Chr 9 was associated with between 2.7% and 3.6% of the total variance and, like the Chr 7 QTL, explained more variance as age increased. The QTL identified on Chr 18 for all BM traits were identified at the suggestive linkage threshold and explained between 2.4% and 3.2% of total phenotypic variance. All QTL identified at the genome-wide significance threshold were robust to changes in the background genetic effects fitted in the model. There was no evidence for the presence of two QTL on any of the chromosomes analyzed.

The likelihood profiles associated the QTL mapped for BM on Chr 8; (A) Mendelian QTL vs. no QTL at 14, 16, and 18 weeks; (B) the three alternative Imprinting models vs. no QTL at 14 weeks.
The magnitude of QTL effects
Positive estimates of genetic effects indicate that the alleles from the MHi line increase the trait, whereas negative effects indicate alleles from the MLi line increase the trait. The distribution of the direction of effect associated with the different QTL shows that increasing alleles originate from both the high and low M-lines. The majority of QTL appear to be additive in action, with no significant dominance deviations. However, the 18-week QTL on Chr 7 is associated with a significant additive effect of 0.146 SD units and negative dominance deviation of −0.237 SD units. The suggestive BM QTL on Chr 2 was associated with large dominance deviations and additive effects which fail to reach significance. For BM at 14 and 16 weeks, the sum of all additive and dominance deviations is close to zero, i.e., −0.118 and −0.106 SD units, respectively. However, for BM at 18 weeks the sum of all QTL additive estimates has a net negative effect of −0.314 SD units, larger than the net effect at 14 and 16 weeks. Net dominance deviations for all BM measurements are close to zero, i.e., −0.029, −0.179, and −0.066 for 14, 16, and 18 weeks, respectively. There was no evidence of the presence of gender × genotype interactions for any of the QTL identified.
Imprinting effects
The single large QTL for BM traits mapped on Chr 8 to 7-8 cM from the most proximal marker was explained by an additive effect that equates to a total effect (2a) between the alternative homozygotes of approximately 2.9 g at 14 weeks declining to 2.6 g at 18 weeks. Analysis of the data to investigate imprinting on the Chr 8 fitting the full Imprinting model (Ppat + Pmat + Pd) indicated that this QTL was associated with a significant (F-ratio = 12.0, 1df, p < 0.05) imprinted effect versus the Mendelian model. The associated QTL positions did not change; however, there was an increase in the percentage variance explained by the QTL to 14.4%, 14.2%, and 12.2% when applying the Imprinting model for 14, 16, and 18 weeks, respectively. Estimates for maternal (imat) and paternal (ipat) imprinting and dominance deviations (d) for BM at 14 weeks were imat = −0.049, ipat = −0.235, and d = 0.044 SD units; at 16 weeks imat = −0.042, ipat = −0.225, and d = −0.010 SD. units; and at 18 weeks imat = −0.058, ipat = −0.211, and d = −0.006. For all estimates only the paternal imprinting effect was significantly (p < 0.05) different from zero. By fitting separate maternal and paternal models, the likelihood profiles suggest the imprinting effect was explained by paternal imprinting (Fig. 2b). No other significant imprinting effects were identified elsewhere in the genome.
Discussion
We have analyzed a single pedigree F2 population with the objective of mapping QTL associated with BM at 14, 16, and 18 weeks. The resultant likelihood profiles suggest five QTL associated with all three BM traits on Chrs 5, 7, 8, 9, and 18; and additional QTL detected at the suggestive threshold for one or more BM traits being identified on six of the remaining autosomes. Because the F2 mapping population was derived from progenitor lines that were not fully inbred (less than 20 generations), we have been able to estimate imprinting effects, which have rarely been explored in QTL mapping studies in mouse populations. The QTL on Chr 8 was explained by paternal imprinting.
One of the most compelling routes to identifying QTL of phenotypic importance is to have QTL confirmed across populations. Chromosomes 1, 2, 4, and 11 have been associated with adult mouse BM, with each loci being replicated in more than seven separate studies (Rocha et al. 2004). Although the QTL mapped in this study are not representative of those detected in previous studies, there is some agreement with previously mapped QTL (Table 3). For example, the QTL mapped to Chr 7 explaining 4.7% of variation in 18-week mass is in agreement with the QTL Bglq6 (Cheverud et al. 1996; Vaughn et al. 1999). In addition, although Bw14 identified by Brockmann et al. (2000) and W10q18 identified by Rocha et al. (2004) fall slightly outside our 1-LOD dropoff confidence interval, this does not take into account their associated confidence intervals, so it may reflect commonality of QTL. Furthermore, the highly significant 16-week mass QTL at the distal end of Chr 5 seems to coincide with the QTL for 6-week mass Bw13 explaining 6% of variance (Brockmann et al. 2000) and Bglq14 associated with late growth mapped by Vaughn et al. (1999). It is feasible that any lack of strong coincidence between QTL mapped at 14-18 weeks and QTL previously identified could be a result of differences in the growth phases measured as previous studies are generally concerned with BM at 10 weeks of age or younger.
While there is some indication of agreement between the QTL mapped in this study and loci previously identified, it is particularly interesting that the most significant QTL on Chr 8 associated with an imprinting effect does not appear to have been identified previously. Although QTL have been mapped to Chr 8, e.g., W10q14 identified by Rocha et al. (2004) explaining 1.3% of phenotypic variance, the magnitude of the QTL effect and location do not appear to agree with the current study. Studies by Anunciado et al. (2001) and Corva et al. (2001) (Bwq3 and Wg3, respectively) identified QTL with locations more distal to the QTL identified in our population. The lack of coincidence could be a result of the M-lines being genetically more distinct from mouse lines used in other QTL mapping studies. However, this hypothesis is not supported by mapping data available from alternative mouse lines derived from the same base population as the M-lines. Using the same progenitor population as the M-lines, divergent selection has been carried out based on protein and later BM producing the P-line (Sharp et al. 1984). QTL for BM were identified on Chr 6 (Christians et al. 2004) and an X-linked QTL of large effect has been identified (Rance et al. 1997) and fine mapped (Liu et al. 2001). Single-marker associations (F2 population of 336, 49 markers) were also investigated on Chrs 1-19 (except 11 and 15) with significant associations on Chrs 2, 3, and 17 (Rance et al. unpublished). These results do not preclude QTL residing elsewhere on the genome of the P-line because the map coverage was poor and information content of some markers was low. However, it is interesting to note that on Chr 8, marker D8Mit31 at 33 cM did not show any marker-associated effects despite an average information content of 0.95 (additive and dominance).
Many of the previous studies mapping QTL associated with BM used progenitor mouse lines with substantially higher divergence in BM. For example, Brockmann et al. (1998) used the high-BM selected DU6 and the unselected DUK lines which differ in BM by 106% at 42 days, and in a recent study by Rocha et al. (2004), lines with a mean BM at 10 weeks of 57.9 g vs. 18.4 g were used to form an F2 population. In the study described here, the progenitor M-lines lines were divergently selected for food intake between 8 and 10 weeks corrected for mean BM, with the objective of keeping both lines at the same BM (Hastings et al. 1997). As a consequence, the relatively unique QTL associated with BM identified in this study may have been anticipated. The M-line selection criteria will prevent independent divergence of QTL of large effect on BM because any change in the frequency of loci within-line associated with increasing BM would need to be balanced with a change of frequency of alleles associated with a decrease in BM so as to maintain an unchanged BM. We postulate that the BM QTL identified in the current study were allowed to diverge as they were associated with pleiotropic effects of genes underlying the selection criterion “food intake at maturity” or other correlated responses associated with energy balance.
The QTL of large effect mapped to Chr 8 is associated with an imprinting effect. The imprinting effect appears to be a result of paternal imprinting as shown when separate maternal and paternal Imprinting models are fitted (Fig. 2b). The QTL was found to be robust to changes in the model fitted, remaining highly significant regardless of the cofactors fitted and gender effect included in the model. Evidence for the imprinting effect did not appear to be influenced by the information content of markers on Chr 8 which were representative of the rest of the genome, i.e., 0.0, 0.20, 0.05, and 0.26 for markers D8MIT141, D8MIT191, D8MIT85, and D8MIT56, respectively, for imprinting and an average of 0.80 for additive and 0.63 for dominance information content. Although the F-ratio of the imprinting versus Mendelian model was sensitive to the imprinting information content at marker D8MIT191 (adjusted by removing data from families where both parents were not homozygous), it is worth noting that the magnitude of imprinting effect remained robust regardless of the information content at both markers D8MIT191 and D8MIT56. All marker loci conformed to the expected Mendelian segregation ratios. The major contribution of paternally inherited alleles is also supported by the QTL estimates obtained from the Imprinting model fitting both maternal and paternal imprinting and dominance effects, where only paternal imprinting estimates are significantly different from zero.
Evidence for an imprinted QTL on Chr 8 suggests that any positional candidate gene proposed should be paternally imprinted. Imprinting is brought about during gametogenesis where parental genomes undergo modification. The result is that genes are differentially expressed when inherited from either the maternal or paternal parent. A key molecular mechanism resulting in imprinting is DNA methylation where imprinted genes are differentially methylated in egg and sperm, and inheritance of these epigenetic marks leads to the differential gene expression (Reik and Walter 2001). To date, around 75 imprinted genes have been identified in the mouse (Beechey et al. 2005) and the distribution of these loci suggests that there are several subchromosomal areas associated with imprinting. The first imprinted genes were not identified until the early 1990s and it is likely that additional imprinted loci remain unidentified.
While a number of loci known to influence growth and development have been shown to be imprinted (including Igf2, Igf2r, Ins1, and Ins2) and shown to cause the imprinting effect associated with identified QTL (IGF2, Van Laere et al. 2003), mouse Chr 8 is not considered to be associated with imprinting (Beechey et al. 2005). However, using comparative mapping, human chromosome locations 13q34 and 8p11-p23 have been associated with reports of imprinting effects (Piantanida et al. 1997; Strichman-Almashanu et al. 2002) although there is conflicting evidence (Jarvela et al. 1998). Despite the lack of any imprinted candidate genes to explain the QTL on Chr 8, Insulin receptor substrate 2 (Irs2) and Adrenergic receptor β-3 (Adrb3) are both good positional candidates for a QTL associated with BM, and it is possible that these genes are imprinted but are as yet unidentified to be so. Knockout mice exist for both Irs2 and Adrb3, and as a result it would be simple to test the hypothesis that these genes were imprinted by making reciprocal crosses between homozygous knockout and wild-type mice. If Irs2 or Adrb3 were imprinted, there would be phenotypic differences between the progeny dependent on the gender of the parent of origin of the wild-type allele. We have not been able to find published evidence to support an imprinting effect for either Irs2 or Adrb3.
Unfortunately, there are no common features present in the protein sequences of imprinted genes to aid identification. However, there are two general features associated with imprinted genes. First, a large proportion of imprinted genes, approximately 88%, are associated with CpG islands compared with an average figure of 47% (Reik and Walter 2001). These CpG islands are believed to be unmethylated in normal cells, with the two exceptions being inactive X chromosome genes and imprinted genes (Strichman-Almashanu et al. 2002). The second feature of imprinted genes is a common occurrence of clustered, direct repeats near to or within the CpG islands. Neumann et al. (1995) proposed that these repeats could be associated with bringing about or maintaining differential methylation. In the study by Strichman-Almashanu et al. (2002), novel imprinted genes were identified using methodologies based on isolation and screening of normally methylated CpG islands. Analysis of the candidate gene Irs2, using the software package CpG Island Explorer (Wang and Leung 2004) identified a large CpG island of 6956 bp with a GC content of 70.1% and CpGobs/CpGexp (observed-to-expected ratio based on GC content) of 0.89 encompassing the entire 3′ exon. Further screening of the CpG islands using the EMBOSS program etandem (website http://www.uk.embnet.org/Software/EMBOSS/) also identified evidence of clustered direct repeats. This might suggest that Irs2 may be imprinted, although it should be noted that these features common to imprinted genes are not fully demonstrative (Reik and Walter 2001). Screening of the candidate gene Adrb3 also identified a CpG island encompassing much of the 3′ area of exon 1; however, no clustered direct repeats were identified. Although CpG islands and clustered direct repeats are associated with imprinted genes, they do not provide conclusive evidence of imprinting. In fact, Yamada et al. (2004) suggests that CpG islands are strongly predictive of methylation but not necessarily imprinting.
To further test the hypothesis that Irs2 and Adrb3 are potentially imprinted candidates, we searched the Candidate Imprinted Transcripts from Expression database (Nikaido et al. 2003). Both candidate genes were not present; however, it is interesting to note that the X-linked Irs4 does show evidence of paternal imprinting.
We have identified QTL for mature BM in a population derived from lines selected for “food intake at maturity.” The loci identified are not representative of QTL for BM identified in previous studies, most notably including a novel paternally imprinted QTL of large effect on mouse Chr 8. Because of the nature of the gene action, screening of candidates for paternal imprinting will provide an additional means of screening positional candidates and may therefore hasten identification of the underlying causative mutations.
This work was supported by Wellcome Grant No. 060086/Z/99/Z. The authors would like to acknowledge the assistance of animal house staff Duncan Wood, Shona Flemming, and Jim Levine for their care of mice. LB is grateful for BBSRC and SEERAD funding part of his work.
References
Anunciado RVP, Nishimura M, Mori M, Ishikawa A, Tanaka S, et al. (2001) Quantitative trait loci for body weight in the intercross between SM/J and A/J mice. Exp Anim 50:319–324
Beechey CV, Cattanach BM, Blake A, Peters J (2005) MRC Mammalian Genetics Unit, Harwell, Oxfordshire. Genetic and Physical Imprinting Map of the Mouse, available at: http://www.mgu.har.mrc.ac.uk/research/imprinting/
Brockmann GA, Haley CS, Renne U, Knott SA, Schwerin M (1998) Quantitative trait loci affecting body weight and fatness from a mouse line selected for extreme high growth. Genetics 150:369–381
Brockmann GA, Kratzsch J, Haley CS, Renne U, Schwerin M, et al. (2000) Single QTL effects, epistasis, and pleiotropy account for two-thirds of the phenotypic F-2 variance of growth and obesity in DIU6i × DBA/2 mice. Genet Res 10:1941–1957
Bunger L, MacLeod MG, Wallace CA, Hill WG (1998a) Direct and correlated effects of selection for food intake corrected for body weight in the adult mouse. Proc 6th World Cong Genet Appl Livestock Product 26:97–100
Bunger L, Renne U, Dietl G, Kuhla S (1998b) Long-term selection for protein amount over 70 generations in mice. Genet Res 72:93–109
Cheverud JM, Rutledge JJ, Atchley WR (1983) Quantitative genetics of development - genetic correlations among age-specific trait values and the evolution of ontogeny. Evolution 37:895–905
Cheverud JM, Routman EJ, Duarte FAM, van Swinderen B, Cothran K, et al. (1996) Quantitative trait loci for murine growth. Genetics 142:1305–1319
Christians JK, Rance KA, Knott SA, Pignatelli PM, Oliver F, et al. (2004) Identification of reciprocal introgression of a QTL affecting body mass in mice. Genet Select Evol 36:577–591
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Coleman DL, Eicher EM (1990) Fat (Fat) and tubby (Tub)—2 autosomal recessive mutations causing obesity syndromes in the mouse. J Hered 81:424–427
Corva PM, Horvat S, Medrano JF (2001) Quantitative trait loci affecting growth in high growth (Hg) mice. Mammal Genome 12:284–290
De Koning DJ, Rattink AP, Harlizius B, Van Arendonk JAM, Brascamp EW, et al. (2000) Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc Natl Acad Sci U S A 97:7947–7950
De Koning DJ, Bovenhuis H, Van Arendonk JAM (2002) On the detection of imprinted quantitative trait loci in experimental crosses of outbred species. Genetics 161:931–938
Deng HW, Deng HY, Liu YJ, Liu YZ, Xu FH, et al. (2002) A genomewide linkage scan for quantitative-trait loci for obesity phenotypes. Am J Hum Genet 70:1138–1151
Doolittle DP, Davisson MT, Guidi JN, Green MC (1996) Catalog of mutant genes and polymorphic loci. In Lyon MF, Rastan S, Brown SDM (eds.) Genetic Variants and Strains of the Laboratory Mouse, (Oxford: Oxford University Press), pp 17–854
Efstratiadis A (1998) Genetics of mouse growth. Int J Dev Biol 42:955–976
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics (Essex, UK: Longman Group)
Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, et al. (1997) Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat Genet 15:269–272
Galton F (1889) Human variety. Nature 39:296–297
GenStat 7 Committee (2003) GenStat Release 7.1 Reference Manual (Oxford, UK: VSN International Limited)
Gong DW, He YF, Karas M, Reitman M (1997) Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta 3-adrenergic agonists, and leptin. J Biol Chem 272:24129–24132
Hahm S, Mizuno TM, Wu TJ, Wisor JP, Priest CA, et al. (1999) Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron 23:537–548
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69:315–324
Haley CS, Knott SA, Elsen JM (1994) Mapping quantitative trait loci in crosses between outbred lines using least-squares. Genetics 136:1195–1207
Hastings IM, Moruppa SM, Bunger L, Hill WG (1997) Effects of selection on food intake in the adult mouse. J Anim Breed Genet 114:419–433
Ishikawa A, Namikawa T (2004) Mapping major quantitative trait loci for postnatal growth in an intersubspecific backross between C57BL/6J and Philippine wild mice by using principal component analysis. Genes Genet Syst 79:27–39
Jansen RC (1993) Interval mapping of multiple quantitative trait loci. Genetics 135:205–211
Jarvela I, Savukoski M, Ammala P, Von Koskull H (1998) Prenatally detected paternal uniparental chromosome 13 isodisomy. Prenatal Diag 18:1169–1173
Jeon JT, Carlborg O, Tornsten A, Giuffra E, Amarger V, et al. (1999) A paternally expressed QTL affecting skeletal and cardiac muscle mass in pigs maps to the IGF2 locus. Nat Genet 21:157–158
Knott SA, Marklund L, Haley CS, Andersson K, Davies W, et al. (1998) Multiple marker mapping of quantitative trait loci in a cross between outbred wild boar and large white pigs. Genetics 149:1069–1080
Lander ES, Green P (1987) Construction of multilocus genetic-linkage maps in humans. Proc Natl Acad Sci U S A 84:2363–2367
Lander ES, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Le Roy I, Perez-Diaz F, Cherfouh A, Roubertoux PL (1999) Preweanling sensorial and motor development in laboratory mice: quantitative trait loci mapping. Dev Psychobiol 34:139–158
Leamy L, Cheverud JM (1984) Quantitative genetics and the evolution of ontogeny .2. Genetic and environmental correlations among age-specific characters in randombred house mice. Growth 48:339–353
Leamy LJ, Pomp D, Eisen EJ, Cheverud JM (2002) Pleiotropy of quantitative trait loci for organ weights and limb bone lengths in mice. Physiol Genomics 10:21–29
Liu XJ, Oliver F, Brown SDM, Denny P, Keightley PD (2001) High-resolution quantitative trait locus mapping for body weight in mice by recombinant progeny testing. Genet Res 77:191–197
MGD (2004) Mouse Genome Database, Mouse Genome Informatics, The Jackson Laboratory, Bar Harbour, ME. Available at http://www.informatics.jax.org/
Nahmias C, Blin N, Elalouf JM, Mattei MG, Strosberg AD, et al. (1991) Molecular characterization of the mouse beta-3-adrenergic receptor - relationship with the atypical receptor of adipocytes. EMBO J 10:3721–3727
Neumann B, Kubicka P, Barlow DP (1995) Characteristics of imprinted genes. Nat Genet 9:12–13
Nikaido L, Saito C, Mizuno Y, Meguro M, Bono H, et al. (2003) Discovery of imprinted transcripts in the mouse transcriptome using large-scale expression profiling. Genome Res 13:1402–1409
Ohlsson H, Karlsson K, Edlund T (1993) Ipf1, a homeodomain-containing transactivator of the insulin gene. EMBO J 12:4251–4259
Piantanida M, Dellavecchia C, Floridia G, Giglio S, Hoeller H, et al. (1997) Ataxic gait and mental retardation with absence of the paternal chromosome 8 and an Idic(8)(P23.3): imprinting effect or nullisomy for distal 8p genes? Hum Genet 99:766–771
Quintanilla R, Milan D, Bidanel JP (2002) A further look at quantitative trait loci affecting growth and fatness in a cross between Meishan and Large White pig populations. Genet Select Evol 34:193–210
Rance KA, Heath SC, Keightley PD (1997) Mapping quantitative trait loci for body weight on the X chromosome in mice. II. Analysis of congenic backcrosses. Genet Res 70:125–133
Rattink AP, De Koning DJ, Faivre M, Harlizius B, Van Arendonk JAM, et al. (2000) Fine mapping and imprinting analysis for fatness trait QTLs in pigs. Mammal Genome 11:656–661
Reed DR, Li X, McDaniel AH, Lu K, Li SR, et al. (2003) Loci on chromosomes 2, 4, 9, and 16 for body weight, body length, and adiposity identified in a genome scan of an F-2 Intercross between the 129P3/J and C57BL/6ByJ mouse strains. Mamm Genome 14:302–313
Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2:21–32
Rice T, Perusse L, Bouchard C, Rao DC (1999) Familial aggregation of body mass index and subcutaneous fat measures in the longitudinal Quebec Family Study. Genet Epidemiol 16:316–334
Rocha JL, Eisen EJ, Van Vleck LD, Pomp D (2004) A large-sample QTL study in mice: I. growth. Mammal Genome 15:83–99
Seaton G, Haley CS, Knott SA, Kearsey M, Visscher PM (2002) QYL Express: mapping quantitative trait loci in of simple and complex pedigrees. Bioinformatics 18:339–340
Selman C, Lumsden S, Bunger L, Hill WG, Speakman JR (2001) Resting metabolic rate and morphology in mice (Mus musculus) selected for high and low food intake. J Exp Biol 204:777–784
Sharp GL, Hill WG, Robertson A (1984) Effects of selection on growth, body composition and food intake in mice. 1. Responses in selected traits. Genet Res 43:75–92
Snyder EE, Walts B, Perusse L, Chagnon YC, Weisnagel SJ, et al. (2004) The human obesity gene map: the 2003 update. Obes Res 12:369–439
Strichman-Almashanu LZ, Lee RS, Onyango PO, Perlman E, Flam F, et al. (2002) A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res 12:543–554
Sun XJ, Wang LM, Zhang YT, Yenush L, Myers MG, et al. (1995) Role of Irs-2 in Insulin and cytokine signaling. Nature 377:173–177
Thomsen H, Lee HK, Rothschild MF, Malek M, Dekkers JCM (2004) Characterization of quantitative trait loci for growth and meat quality in a cross between commercial breeds of swine. J Anim Sci 82:2213–2228
Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, et al. (2003) A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425:832–836
van Ooijen JW (1992) Accuracy of mapping quantitative trait loci in autogamous species. Theoret Appl Genet 84:803–811
Vaughn TT, Pletscher LS, Peripato A, King-Ellison K, Adams E, et al. (1999) Mapping quantitative trait loci for murine growth: a closer look at genetic architecture. Genet Res 74:313–322
Visscher PM, Thompson R, Haley C (1996) Confidence intervals in QTL mapping by bootstrapping. Genetics 143:1013–1020
Wang L, Leung FC (2004) An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics 20:1170–1177
Yamada Y, Watanabe H, Miura F, Soejima H, Uchiyama M, et al. (2004) A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q. Genome Res 14:247–266
Yang RC (2004) Epistasis of quantitative trait loci under different gene action models. Genetics 167:1493–1505
Zeng Z-B (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rance, K.A., Fustin, JM., Dalgleish, G. et al. A paternally imprinted QTL for mature body mass on mouse Chromosome 8. Mamm Genome 16, 567–577 (2005). https://doi.org/10.1007/s00335-005-0012-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-005-0012-4