
Abstract
The set of LXS recombinant inbred (RI) strains is a new and exceptionally large mapping panel that is suitable for the analysis of complex traits with comparatively high power. This panel consists of 77 strains—more than twice the size of other RI sets— and will typically provide sufficient statistical power (β = 0.8) to map quantitative trait loci (QTLs) that account for ∼25% of genetic variance with a genomewide p < 0.05. To characterize the genetic architecture of this new set of RI strains, we genotyped 330 MIT microsatellite markers distributed on all autosomes and the X Chromosome and assembled error-checked meiotic recombination maps that have an average F2-adjusted marker spacing of ∼4 cM. The LXS panel has a genetic structure consistent with random segregation and subsequent fixation of alleles, the expected 3–4 × map expansion, a low level of nonsyntenic association among loci, and complete independence among all 77 strains. Although the parental inbred strains—Inbred Long-Sleep (ILS) and Inbred Short-Sleep (ISS)—were derived originally by selection from an 8-way heterogeneous stock selected for differential sensitivity to sedative effects of ethanol, the LXS panel is also segregating for many other traits. Thus, the LXS panel provides a powerful new resource for mapping complex traits across many systems and disciplines and should prove to be of great utility in modeling the genetics of complex diseases in human populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Recombinant inbred (RI) strains have been used in genetic mapping for approximately 30 years (Bailey 1981; Taylor 1978). They provide numerous advantages, one of the foremost being that strains retain their genetic identity for hundreds of generations but only need to be genotyped once. Furthermore, the additive genetic variance in a panel of RI strains is twice that of a corresponding F2 intercross for the same trait, a feature that increases the effective power to map many types of subtle polygenic traits. (Dominance effects can be readily estimated by making sets of isogenic F1 hybrids among RI strains.) Recombination rates in RI strains also average fourfold higher than in conventional test crosses (Haldane and Waddington 1931; Lynch and Walsh 1998; Williams et al. 2001), resulting in improved mapping resolution compared with backcrosses or intercrosses. It is possible to treat, test, and measure many isogenic individuals and to detect and reduce both environmental and technical sources of variance. This attribute is typically exploited to increase the precision of strain means, boosting the effective heritability of a trait. In fact, the effective heritability for genetically specified traits asymptotically approaches 100% with increased sample size (Belknap 1998). This is a crucial advantage for mapping fitness-related or behavioral traits and molecular phenotypes such as transcript and protein expression levels that often have low heritability and high environmental liability.
By phenotyping strains rather than individual mice, it is also possible to systematically explore a crucial and still uncharted area of mouse genetics, namely, gene-by-environment interactions. Sets of loci that modulate a trait under one set of conditions and environment may differ markedly from the set of loci that control a trait under a second set of conditions and environment. By using RI strains, it becomes practical to intentionally vary environmental factors and to study, for example, the effects of stress, environmental enrichment, and diet on behavioral traits or disease susceptibility.
Perhaps the most significant advantage of using RI strains is that they are a renewable resource for long-term interdisciplinary and collaborative research. Thousands of different phenotypes can be measured at different times and in different laboratories on the same genotypes, using the same strains. This makes it possible to explore genetic correlations, gene pleiotropy, and interactions among traits and the quantitative trait loci (QTLs) modulating those traits. The cumulative nature of such studies is well illustrated by the BXD RI strains, where over 50,000 traits have been collected in this one panel (Chesler et al. 2003; http://www.webqtl.org/search.html).
The most serious disadvantage of RI strains has been the modest numbers of strains available per set. While plant geneticists typically use 100–1000 RI lines (e.g., Stenehjem and Bruggemann 2001) to map traits, mouse and rat geneticists have been limited to sets of 15–30 strains. This has greatly diminished enthusiasm for the use of RI strains except among behavior geneticists. The power to detect a QTL increases with increasing number of RI strains (Belknap 1998; Belknap et al. 1996), and with only 26 strains (the size of the original BXD panel) a QTL must have an unusually large effect (56% of the additive genetic variance, V A) to be detected with a power of 0.8 without subsequent confirmation (Belknap et al. 1996). The simple solution to this problem is to generate larger panels of RI strains from genetically diverse parental strains.
In this article, we provide an initial genetic characterization of the LXS panel of RI strains. This panel currently consists of 77 inbred strains that trace their descent from the inbred progeny of a multigeneration cross of eight diverse laboratory strains begun by Gerald McClearn and colleagues 45 years ago (McClearn et al. 1970). We estimate that the LXS RI panel collectively incorporates approximately 3600 recombination breakpoints, providing subcentimorgan precision for monogenic traits and 1–10-cM precision for QTLs modulating complex polygenic traits. We have genotyped the panel using a set of 330 well-characterized and physically mapped MIT microsatellite loci. Our maps provide good and highly usable, but not yet comprehensive, coverage. Given the genetic diversity of the source strains, this RI panel should have widespread utility in dissecting polygenic traits and in modeling many complex human diseases.
Materials and methods
Animals
The LXS RI panel was generated (Fig. 1) in the specific pathogen-free facility at the Institute for Behavioral Genetics, Boulder, CO, from crosses between the ILS (Inbred Long Sleep, abbreviated to L) and ISS (Inbred Short Sleep, abbreviated to S) strains of mice (Markel et al. 1995). These two strains are the product of bidirectional selection for long or short duration of loss-of-righting response following a high-dose, intraperitoneal injection of ethanol. The ILS and ISS and their progenitors have been widely used in alcohol research, and they have been cited in approximately 400 publications over a 30-year period. The LXS panel was initiated at the direction of one of the authors (JCD) and maintained and systematically inbred, until funding was obtained for their continued analysis and inbreeding by TEJ and BB.

Derivation of the LXS RI strains showing the original 8-way cross, selection for differential ethanol sensitivity, and inbreeding. Approximately equal numbers of reciprocal F1 crosses between ILS and ISS were started in 1996, and currently, after inbreeding is complete, approximately equal numbers remain. Each of the four F2 crosses generated a unique pairing of Y Chromosome and maternal (mitochondrial) contribution.
The original derivation of the ILS and ISS strains has been described in detail by Bennett and colleagues (2002) but, in brief, these strains trace their descent from a heterogeneous stock (McClearn et al. 1970; McClearn and Kakihana 1981) derived from an eight-way cross of inbred strains maintained at that time by the Cancer Research Genetics Laboratory at the University of California at Berkeley (A, AKR, BALB/c, C3H/2, C57BL, DBA/2, Is/Bi, and RIII). To produce the LXS RIs, ILS and ISS mice were reciprocally intercrossed (three matings of each cross) and all four reciprocal matings (ten mating pairs per cross) of F1 mice were used to produce the F2 generation. Inbreeding at the F2 generation began with 125 mating pairs derived in approximately equal numbers from the four matings used to produce the F2 (Fig. 1). The F2 crosses were designed to generate all possible combinations of Y Chromosome (L or S) and mitochondrial genomes (L or S) in approximately equal numbers of strains in all four combinations.
In subsequent generations, three brother–sister matings per strain were set up whenever possible. Matings were maintained until 2–3 litters had matured and the next generation of filial (F) matings had been initiated. A mating was considered unproductive if no litter was produced within 60 days, or if all mice died within a few days of birth. In the case of unproductive matings, progeny from the same line but previous generation were used to replace the original mating, if available; if not, males were switched between existing matings. The 20th generation of inbreeding was completed for the last of the LXS strains in 2002. Currently, four mating pairs are maintained per strain. The number of consecutive filial matings currently ranges from F22 to F25.
Genotyping
DNA purification and microsatellite marker screening were done as described by Williams et al. (2001), Lu et al. (2001), and Bennett et al. (2002). Genotyping was begun at F14–18, a stage at which inbreeding was estimated to be ∼94% complete. DNA was often pooled from two mice per strain to maximize the probability of detecting unfixed loci. Such loci were genotyped again, in subsequent generations, though not always with pooled DNA samples. We also routinely regenotyped markers associated with flanking proximal and distal recombination events in single strains (so-called double recombinants) to further reduce possible genotyping errors.
Databases and analytical tools
Marker genotypes were entered directly into a relational database system (FileMaker Pro) and exported to Microsoft Excel, MapManager QTX (Manly and Olson 1999; http://mapmgr.roswellpark.org/mmQTX.html), and R/qtl (Broman et al. 2003; http://www.biostat.jhsph.edu/∼kbroman/qtl/) for subsequent analysis. We also assembled a database on the physical map location of just over 6300 MIT microsatellite loci by BLAT analysis of MIT sequence against the Mouse Genome Sequencing Consortium build of the mouse genome at http://www.genome.ucsc.edu. This made it possible to order markers using either the February 2002 release of the physical assembly (http://www.genome.ucsc.edu/cgi-bin/hgGateway) or the strain distribution patterns themselves. We generally followed the order of MGSCv3, but since this assembly is still incomplete, in some instances our genetic maps strongly support an alternate marker order. In most cases of contradictory marker orders, we weighted the physical information most strongly but looked for consensus involving that order and one which minimized apparent double-recombination events. Genotype tables for the LXS strains and additional analyses can be obtained as supplemental material at two sites: (1) WebQTL (http://www.webqtl.org) and (2) as a set of text, Excel, FileMaker, QTX, and R/qtl files at ftp://atlas.utmem.edu/public/lxs [inactive]. These files include both the known or putative physical location of each marker, its centimorgan (cM) position in the final iteration of the Chromosome Committee Reports in 2001 (http://www.informatics.jax.org/ccr/searches/about.cgi?year=2001), and the cM marker positions calculated from the set of LXS strains themselves.
Results and discussion
Strains
Construction of the LXS panel was initiated by John DeFries and colleagues in 1996, with 125 F2 mating pairs from reciprocal LXS and SXL F1 matings. At the 14th generation 86 lines survived, and, currently, at the 22nd generation 77 strains survive. Compared to the loss of 5 of 12 strains of the first RI panel (Bailey 1971) or Green’s (1966) estimate that only 1 in 6 strains survive 20 generations of inbreeding, the survival of 77 of 125 inbred strains was a better outcome than expected.
Genotyping
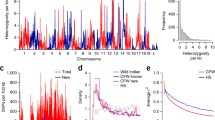
We initially screened 813 MIT microsatellite markers for sequence length polymorphisms between the parental strains ISS and ILS. Of this set, 361 (44%) were polymorphic using methods that consistently detect a difference of 4 bp or greater and that rarely detect differences of as little as a single repeat (2 bp). This level of genetic difference is similar to that observed in other inbred strains. We subsequently genotyped 330 of these markers, which were comparatively evenly distributed across the genome, in all of the RI strains (Fig. 2). A total of 172 strain–generation combinations were typed and retyped, approximately 80,000 genotypes in total. Here we report on the final set of genotypes for these 330 MIT markers, in a final set of 73 strains. Four strains that were apparently not fully inbred (see below) are still being genotyped. If the 330 markers were spaced evenly across the mouse genome, this would be equivalent to mean separation of ∼4.1 cM [1450 cM / (330 + n of chromosomes)]. The median and mean distance between markers (excluding the coverage gap at the ends of the chromosomes) was similarly 4–5 cM (median ∼4.0 cM, mean ∼4.7 cM, Fig. 2). Although the maximal gap on each chromosome is still large and quite variable (7–23 cM; Table 1), a randomly selected gene will typically be well under 4 Mb from the closest flanking marker. Thus, the map, while not final, is nonetheless quite usable for the analysis of both Mendelian and complex traits.

Location of markers (horizontal bars) used to generate the LXS genetic map, consisting of 330 polymorphic microsatellite markers. The largest single gap on each chromosome is described in more detail in Table 1. These marker positions are based on the LXS genetic map; the proximal end of the chromosome is shown at the 0 position; the location of the distal end of the chromosome was determined by using the MGD distance between the last distal marker genotyped and the last marker position on that chromosome.
Comparison of genotypes between DNA samples collected at F14–18 and at F18–20 identified a number of apparently unfixed or miscalled loci. Most of these genotypes were retested at F22–23. Four strains showed a large number of inconsistent genotypes over the multiple generations of testing. These strains (LXS60, LXS102, LXS107, and LXS115) are still unfixed in a number of intervals and are therefore undergoing additional genotyping and inbreeding. These strains have been excluded from the following results and discussion.
Marker fixation
The percent of markers segregating decreases by approximately 19.1% in each successive generation of full sib mating (Green 1981). By F20. 1.8% of loci (6 of 330) are expected to be unfixed (Fig. 3A). In fact, only one strain greatly exceeded this value, with 17 segregating markers. Most strains had 1%–2% of markers still segregating (Fig. 4B: mean = 1.1% at F20 versus the expectation of 1.8%), and in 22 strains we found no unfixed markers (Fig. 3B). Since we did not invariably type DNA samples from both parents that gave rise to the next generation of inbreeding, the percentage of unfixed alleles will tend to be underestimated slightly. But even if we double the number of loci segregating (the most conservative estimate of total residual unfixed loci is 2× the number of detected unfixed loci), then we have still achieved 98% fixation at the F20. The RI strains are now 2–5 generations beyond the F20 and thus are estimated to have fewer than 1 in 100 unfixed loci.

Unfixed loci. A. Percent of loci (± SE) segregating at each generation of sampling. The expectation for the percent of unfixed loci resulting from full-sib crosses is indicated by the open circles along with the observed value for all generations for which DNA was tested. Fewer strains were sampled at the earlier generations (e.g., 13 and 14), so these values are not representative. B. Percent loci segregating at the second genotyping, F14–18. There were 43 strains that had significantly (p < 0.05) fewer than the expected 17 (5%) unfixed marker loci.

The distribution of the ILS allele frequency computed for (A) each of the 330 markers genotyped in the LXS panel with normal curve superimposed, and (B) each individual strain. For any given marker or strain, 50% of the alleles in the RI panel were expected to be from ILS and 50% from ISS (horizontal line).
Segregation distortion
Segregation distortion in RI sets can give rise to serious problems in mapping QTLs. In particular, an imbalance of L and S alleles at unlinked markers on different chromosomes generated by epistatic interactions that affect fecundity or fitness could lead to spurious nonsyntenic associations (Williams et al. 2001). It is straightforward to test for deviations of allele segregation. At any given marker, the expectation is that half of the LXS strains should carry an ILS allele and the other half should carry an ISS allele. In the absence of selection or breeding errors, deviations around this 50:50 expectation should have a binomial distribution. The observed frequency of these parental alleles was very close to expectation. The modal value, as shown in Figure 4A, is 50% (mean ± SD = 49.0 ± 0.6%). This distribution also did not deviate significantly from the binomial prediction (observed χ2 =12.62; χ 20.95 = 14.07, p ∼ 0.085; Fig. 4). The extremes were LXS49 and LXS10 with 27.6% and 63.6% ILS marker genotypes, respectively.
Eleven markers in three chromosomal regions showed more ILS alleles than expected (p = 0.017, pointwise test), although none of these deviations was statistically significant following Bonferroni correction. As Bonferroni corrections are probably overly conservative (see below), these regions are discussed briefly here, as they may represent areas of selection. In addition, any nonsyntenic associations that arise as a result of segregation distortion can affect subsequent mapping. On Chr 11, eight markers between 46 and 66 cM had an average of 68% ILS alleles per marker, differing from expectation at p = 0.025 (pointwise). On Chr 6, three markers from the proximal end to 15.6 cM also showed an excess of ILS alleles, averaging 67% (p < 0.025). One of these markers, D6Mit341 at 7.5 cM, showed a nonsyntenic correlation with a marker on Chr 2, possibly because of the excess of ILS alleles, although the Chr 2 marker, D2Mit81, did not show an equivalent distortion. On Chr 7, six of eight markers between 41 and 60 cM showed an excess of ISS alleles. The Chr 7 region also is characterized by a significant excess of the albino genotype (55 of 77 strains; χ 21 = 14.1, p = 0.002). (This excess is not significant with a Bonferroni correction for 250 or more tests.) As 330 markers are not 330 independent linkage groups, a more realistic, though still conservative, Bonferroni correction might be 100, over twice the average number of breakpoints per strain (2.14). This correction suggests that the bias on Chr 7 may be statistically significant and may represent either an inadvertent bias in selecting white pups for mating during the generation of the LXS panel or, more likely, a fitness advantage of an S allele that is linked to the albino c allele at the Tyr locus carried by ISS. Thus, the Chr 7 region, and perhaps the other regions, represent areas of selection, either inadvertent or fitness-related.
Marker distribution
Despite our best attempt, the spacing of genetic markers was not uniform across the genome (Fig. 2). The average distance between markers is 7.4 Mb (this figure was calculated by dividing the total physical distance from the proximal marker to the distal marker on each chromosome, summed over all chromosomes, divided by 330 markers) or roughly 4 cM. Although the variability is large, as shown by the right hand tail in Figure 5, which shows the distribution of intermarker gaps, there are relatively few gaps larger than 16 Mb. The largest gap on each chromosome is given in Table 1; these gaps were identified using the LXS map, based on recombinations observed in the LXS RI, since the utility of this panel for mapping is based in part on this recombination frequency. These gaps are described using two additional sources in Table 1: (1) MGD positions based on consensus marker locations from the Mouse Genome Informatics Web Site, The Jackson Laboratory, Bar Harbor, ME, December 2003 (URL: http://www.informatics.jax.org); and (2) the physical distance between the flanking markers in megabases (Mb) based on the UCSC Genome Brower and NCBI database (http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid=10090). The largest regions between markers on each chromosome range from 9.3 to 43.4 Mb (Table 1). The two largest gaps, on Chromosomes 4 and X, are indicated by superscript “f” Table 1 and are shown in the histogram of gap sizes in Figure 5. In only one case (Chromosome 17) is there a larger gap than indicated in Table 1, between the last marker shown in Figure 2 and the distal end of the chromosome.

Histogram of interval length between microsatellite markers in Mb, based on the marker positions from the NCBI database (http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid = 10090). The two largest gaps have a superscript “f” in Table 1 and are discussed in the text.
Some of these gaps may be recombinational hotspots. For example, on Chr 6, the LXS map distance between D6Mit291 and D6Mit15 is 38.3 cM but the physical distance is only 9.3 Mb (Fig. 2). Similarly, on Chromosome 18, the LXS map distance between D18Mit7 and D18Mit128 is 66.2 cM, while the physical distance is 12.1 Mb (Fig. 2). Other gaps are probably due to mapping discrepancies in the Mouse Genome Database (http://www.jax.org/). For example, the genomic sequence (NCBI) suggests that the markers mapping to and defining the distal end of Chr 2 (Fig. 2) are actually proximal to our most distal marker in the LXS panel, indicating that the large terminal gap shown in Figure 2 is not real. The majority of the gaps (e.g., the two largest gaps on Chr 4 and Chr X; Fig. 5 and Table 1) appear to be real, as their physical sizes (41.3 and 43.4 Mb, respectively) approximate the genetic map distance reported by MGD and observed in the LXS.
Other regions may simply not be polymorphic due to shared ancestry in the mouse strains used to generate the common inbred strains (Wade et al. 2002). Moreover, the ILS and ISS parental strains share chromosomal-regions that are expected to be identical by descent (IBD) at multiple sites throughout their genome. Given that eight inbred strains (six of which were commonly used strains) contributed to the heterogeneous stock (HS) from which the LS and SS selected lines were derived (McClearn and Kakihana 1981; Fig. 1), one-eighth of the genome should be IBD, assuming random fixation. In fact, this figure is probably an underestimate due to fixation of alleles in the HS progenitor strains. In 15 of the gap regions (Table 1, last 2 columns), 70% or more of the markers tested were not polymorphic, consistent with the hypothesis that some or all of these regions are IBD. Although in most gaps exhaustive sampling of MIT markers was not done, on Chromosomes 3, 8, and 11, we genotyped 83%, 56%, and 61%, respectively, of all microsatellite markers listed at MGD. We also typed all available MIT microsatellites in two intervals which appear to be IBD. On Chr 2, none of 21 MIT markers between 147.0 and 160.3 Mb was polymorphic (Bennett et al. 2002), and on Chr 15, none of 19 markers between 70.0 and 72.9 Mb was polymorphic (unpublished).
The order of markers (Fig. 2) was generally consistent with the MGSCv3 physical map. In five cases, on four chromosomes, the LXS order appeared to reverse the physical map order. (There is some uncertainty as to the position of D15Mit93 on the physical map so this reversal may not be real.) In all of these cases, there was some ambiguity in the genetic maps, with some supporting the physical map and others supporting the LXS genetic map. In the most extreme example, no genetic map supports the marker order switch involving D14Mit257 and D14Mit101.
Map expansion
The mean uncorrected recombination map length of the LXS RI strains is 41.5 Morgans (M) compared with the 13–15-M lengths of single-generation meiotic maps. This represents a map expansion of about 3.4-fold, with a 95% confidence interval (CI) of 3.1–3.7. This expansion was calculated as the ratio of the expanded map distance, between proximal and distal markers, to the unexpanded map distance, from MGD, summed over all chromosomes. Map expansion in F2-derived RI strains is expected to average four-fold in RI strains mapped with infinite marker density (Haldane and Waddington 1931), 3.7-fold with 1-cM marker density, and two-fold with 16-cM marker density (Williams et al. 2001). Therefore, the 3.4-fold expansion we observed based on a 4–5-cM map density is in line with expectation. The map expansion among strains ranged from a low of 2.2-fold in LXS3 to a high of 4.4-fold in LXS35 (Fig. 6).

Mean map expansion in LXS RI strains (3.39 ± 0.15), expressed as mean number of recombinations per 100 cM. This factor was calculated by dividing the total number of recombinations per strain by the total map distance between the most proximal and most distal markers, summed over all chromosomes, in the 73 strains used for mapping.
As expected, all chromosomes showed expansion in total length (Table 1), as determined by summing the distances (based on MGD and recombination frequency for LXS) between the extreme centromeric and telomeric markers. Thus, each value in Table 1 slightly underestimates the chromosome length. The average distance between pairs of adjacent markers based on the LXS map is 13.2 cM; the equivalent distance on the MGD map is 4.3 cM. Most intervals (94%) between marker pairs (311) are expanded in LXS relative to MGD. However, 35 intervals were contracted, and one showed no change. Most of the apparent contractions are quite small, less than 3 cM, and only two of these intervals fall outside of a two-SD cutoff from the mean size. These two intervals, one on Chr 16 at approximately 50 cM, and the second on the X Chr at approximately 58 cM, are not supported by the physical map and thus are probably not real.
Independence of strain genotypes
If RI strains share large regions with similar or identical genotypes across a number of chromosomes, this is generally caused by breeding errors. Nonindependence of strain genotypes reduces the information content of an RI set and distorts genetic maps. To identify and exclude such nonindependent strains, we computed all pairwise correlations of genotypes of LXS strains. Figure 7 shows the genetic similarity of the RI strains to each other based on the percentage of identical genotypes at 330 markers. This analysis revealed that two of the LXS strains were 97% identical, indicating that they were not independent. One of these two strains has been removed and is not counted as one of the 77 strains comprising the LXS panel. The normal distribution of genotypic similarity for the 77 remaining strains indicates that the percent identities are randomly distributed as expected, and thus all 77 strains are informative for genetic mapping. The slight skew gave a mean of 50.5% identity, but this deviation was not statistically significant. The distribution range also matches closely with the BXN RI strains characterized for strain independence by Williams et al. 2001) and the HXB/BXH rat RI panel characterized by Jirout et al. (2003).

Genetic identity among LXS RI strains. The percentage of identical genotypes was determined for all pairwise combinations of 78 strains from a matrix of strain genotypes. One pair of strains showed 97% identity (*) and one of those strains was removed from the panel.
Nonsyntenic correlations
Two markers that are tightly linked will have nearly the same strain distribution pattern (SDP) and a high positive correlation. In contrast, unlinked markers and markers on two different chromosomes in general should not have significant positive or negative correlations beyond that expected by chance. However, nonsyntenic associations arise when regions on different chromosomes show statistically significant positive or negative correlations. This can be due to chance fixation of the same alleles, but in some cases it is likely due to selective advantage of epistatic combinations of alleles on two or more different chromosomes (Williams et al. 2001). Nonsyntenic associations are revealed by searching a correlation matrix of marker genotypes for all pairs of markers with high correlations. A number of nonsyntenic loci are effectively linked, albeit weakly. One such correlation (r = 0.33) between D6Mit341 and D2Mit81 results from a previously mentioned disproportionately high representation of ILS alleles at both markers. Given the very large number of correlations we examined (330 × 329/2), we expect some correlations with statistically significant absolute values by chance. The number of effective tests is much less than 54,285 as a result of linkage, but even if we assume the effective number of tests is approximately 10,000, then a correlation of 0.3 is still not significant at p < 0.05. Nevertheless, even though this effect is not statistically significant, it will still generate weak spurious correlations. In general, the large size of the LXS panel makes it far less sensitive to nonsyntenic association than the BXD RI set (n = 26) and even smaller panels such as the CXB set (n = 13).
Genetic mapping in the LXS
We are presently using the LXS RI panel to map a variety of alcohol-related traits, including ethanol-induced loss of righting, psychomotor activation, hypothermia, blood ethanol concentration at awakening, and acute functional tolerance, and numerous CNS morphometric traits. These ethanol-related traits in particular are all known to differ significantly between the ILS and ISS parental strains (Markel et al. 1995; Owens et al. 2002). Many other traits have been shown to differ between the selected, noninbred LS and SS strains and have been mapped in a previous, smaller RI panel (LSXSS; DeFries et al. 1989). A brief sampling of these traits includes the alcohol-related phenotypes listed above, as well as ethanol preference (Church et al. 1979), corticosterone release in response to acute (Wand 1990) or chronic ethanol treatments (Wand 1989), a variety of behavioral stressors (Wand 1990), structural responses in the hippocampus in response to acute (Poelchen et al. 2000) or chronic ethanol (Lang et al. 1997; Markham et al. 1987; Huang and McArdle 1993), open field activity and defecation (Swanberg et al. 1979), and the anxiolytic effects of ethanol measured in several different tests (Allan and Isaacson 1985; Cao et al. 1993; Lapin et al. 1996; Stinchcomb et al. 1989). In addition, differential sensitivity to numerous other anesthetic agents, alcohols, and drugs of abuse have been found in the ILS and ISS (Simpson et al. 1998). We therefore anticipate that the large LXS panel will find extensive uses in neurogenetic, neuropharmacological, and behavioral studies (cf. Deitrich and Pawloski 1990).
Other phenotypes, such as body weight, and the response of body temperature to dietary restriction do not differ between the parental strains but nonetheless show significant variability and heritability across the LXS strains and, thus, should also be particularly amenable to QTL analysis (Rikke et al. 2003, 2004; Bennett et al. unpublished). We believe this panel will thus provide a valuable new resource to the mouse community for genetic mapping. In a panel of 77 RIs, QTLs accounting for approximately 25% of V A can be detected at a statistically significant p-value (<0.0001) with a power of 80%, by testing replicate mice within each strain
The exact within-strain sample size depends on the heritability of the trait (Belknap 1998). In a simple illustration of the increased power in the LXS panel, the tyrosinase locus (Tyr) was mapped in this panel, and a single major peak at D7Mit350 (73.3 Mb) near the Tyr locus (75.9 Mb) was identified with a LOD of 29. In an earlier RI panel (LSXSS) of 27 strains, derived from the noninbred, selected LS and SS lines (DeFries et al. 1989), Tyr mapped to the same position with a LOD of 12. The albino LOD-score analysis was extended to 19 other loci as well, and a similar improvement of 2.8-fold in LOD score was obtained on average when using 73 strains versus 27 strains. In general, LOD scores are directly proportional to the number of strains (n): LOD ≈ (n × r 2)/4.61, where r 2 is the QTL effect size, with 1 df. Several of the alcohol-related phenotypes that we are currently investigating in the LXS panel have previously been assessed in the LSXSS (Markel et al. 1996; Erwin et al. 1997; Gehle and Erwin 2000); we are currently mapping these traits in the LXS panel and anticipate similar improvements in significance of QTLs.
Conclusions
We have generated and characterized the LXS panel of 77 RI strains that will be a useful mapping resource for the mouse genetics community. Currently, 330 microsatellite marker genotypes at an average marker spacing of 4–5 cM have been assessed in 73 of 77 strains. We are adding additional markers to the SDP to achieve a smaller intermarker distance and are completing genotypic assessment of the four strains with inconsistent genotypes in the earlier screens. The number of strains in this panel provides 80% power, without subsequent confirmation, to map QTLs that explain as little as 23% of V A at a statistically significant level. Even at the present marker density, the mapping precision will be improved by approximately threefold over a panel of 27 strains.
References
A Allan R Isaacson (1985) ArticleTitleEthanol-induced grooming in mice selectively bred for differential sensitivity to ethanol Behav Neural Biol 44 386–392 Occurrence Handle1:CAS:528:DyaL28Xks12qsQ%3D%3D Occurrence Handle4084184
DW Bailey (1971) ArticleTitleRecombinant inbred strains Transplantation 11 325–327 Occurrence Handle1:STN:280:CS6B2M3ptVE%3D Occurrence Handle5558564
DW Bailey (1981) Recombinant inbred strains and bilineal congenic strains HL Foster JD Small JG Fox (Eds) The Mouse in Biomedical Research Academic Press New York 223–239
JK Belknap (1998) ArticleTitleEffect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains Behav Genet 28 29–38 Occurrence Handle10.1023/A:1021404714631 Occurrence Handle1:STN:280:DyaK1c3jsVOhtQ%3D%3D Occurrence Handle9573644
JK Belknap SR Mitchell LA O’Toole ML Helms JC Crabbe (1996) ArticleTitleType 1 and type 2 error rates for quantitative trait loci (QTL) mapping studies using recombinant inbred mouse strains Behav Genet 26 149–160 Occurrence Handle1:STN:280:BymB3cvit10%3D Occurrence Handle8639150
B Bennett M Beeson L Gordon TE Johnson (2002) ArticleTitleReciprocal congenics defining individual quantitative trait loci for sedative/hypnotic sensitivity to ethanol Alcohol Clin Exp Res 26 149–157 Occurrence Handle1:STN:280:DC%2BD383isFOgtw%3D%3D Occurrence Handle11964553
KW Broman LB Rowe GA Churchill K Paigen (2002) ArticleTitleCrossover interference in the mouse Genetics 160 1123–1131 Occurrence Handle11901128
W Cao T Burkholder L Wilkins AC Collins (1993) ArticleTitleA genetic comparison of behavioral actions of ethanol and nicotine in the mirrored chamber Pharmacol Biochem Behav 45 803–809 Occurrence Handle10.1016/0091-3057(93)90124-C Occurrence Handle1:CAS:528:DyaK3sXlvFOlsLo%3D Occurrence Handle8415818
EJ Chesler JB Wang L Lu Y Qu KE Manly et al. (2003) ArticleTitleGenetic correlates of gene expression in recombinant inbred strains: A relational model system to explore neurobehavioral phenotypes Neuroinformatics 1 343–357 Occurrence Handle10.1385/NI:1:4:343 Occurrence Handle15043220
AC Church JL Fuller L Dann (1979) ArticleTitleAlcohol intake in selected lines of mice: Importance of sex and genotype J Comp Physiol Psychol 93 242–246 Occurrence Handle1:STN:280:CSaB3Mnptl0%3D Occurrence Handle457948
JC DeFries JR Wilson VG Erwin DR Petersen (1989) ArticleTitleLS × SS recombinant inbred strains of mice: Initial characterization Alcohol Clin Exp Res 13 196–200 Occurrence Handle1:STN:280:BiaB2s%2FptlY%3D Occurrence Handle2658655
Deitrich RA, Pawlowski AA (1990) Initial sensitivity to alcohol. In: A Workshop on Alcohol Intoxication (Keystone, CO: U.S. Department of Health and Human Services, Public Health Service)
VG Erwin PD Markel TE Johnson VM Gehle BC Jones (1997) ArticleTitleCommon quantitative trait loci for alcohol-related behaviors and central nervous system neurotensin measures: Hypnotic and hypothermic effects J Pharmacol Exp Ther 280 911–918 Occurrence Handle1:CAS:528:DyaK2sXivVWqsrc%3D Occurrence Handle9023306
VM Gehle VG Erwin (2000) ArticleTitleThe genetics of acute functional tolerance and initial sensitivity to ethanol for an ataxia test in the LS × SS RI strains Alcohol Clin Exp Res 24 579–587 Occurrence Handle10.1097/00000374-200005000-00001 Occurrence Handle1:CAS:528:DC%2BD3cXktFGiurs%3D Occurrence Handle10832898
EL Green (1981) Genetics and Probability in Animal Breeding Experiments Oxford University Press New York
JBS Haldane CH Waddington (1931) ArticleTitleInbreeding and linkage Genetics 16 357–374 Occurrence Handle1:STN:280:Bi2B3c%2FotVM%3D Occurrence Handle7342407
GJ Huang JJ McArdle (1993) ArticleTitleChronic ingestion of ethanol increases the number of Ca2+ channels of hippocampal neurons of long-sleep but not short-sleep mice Brain Res 615 328–330 Occurrence Handle10.1016/0006-8993(93)90044-N Occurrence Handle1:CAS:528:DyaK3sXltlegsr4%3D Occurrence Handle8395960
M Jirout D Krenova V Kren L Breen M Pravenec et al. (2003) ArticleTitleA new framework marker-based linkage map and SDPs for the rat HXB/BXH strain set Mamm Genome 14 537–546 Occurrence Handle10.1007/s00335-003-2266-z Occurrence Handle1:CAS:528:DC%2BD3sXmslCqt74%3D Occurrence Handle12925886
D Lang M Beno E Fifkova H Eason (1997) ArticleTitleFine structure of hippocampal dendrites in the dentate fascia of LS/SS mice after chronic ethanol treatment Prog Neuropsychopharmacol Biol Psychiatry 21 1031–1042 Occurrence Handle10.1016/S0278-5846(97)00096-1 Occurrence Handle1:CAS:528:DyaK2sXmtVGjsb0%3D Occurrence Handle9380786
IP Lapin S Mirzaev (1996) ArticleTitleThe contrary effects of ethanol on the behavior of short-and long-sleep C57BL/6 mice in a dark-light chamber Eksp Klin Farmakol 59 50–52 Occurrence Handle1:CAS:528:DyaK28XmtVWgtbw%3D Occurrence Handle8974567
L Lu DC Airey RW Williams (2001) ArticleTitleComplex trait analysis of the hippocampus: Mapping and biometric analysis of two novel gene loci with specific effects on hippocampal structure in mice J Neurosci 21 3503–3514 Occurrence Handle1:CAS:528:DC%2BD3MXjs1ant7Y%3D Occurrence Handle11331379
M Lynch B Walsh (1998) Genetics and Analysis of Quantitative Traits Sinauer Associates, Inc. Sunderland, MA
KF Manly JM Olson (1999) ArticleTitleOverview of QTL mapping software and introduction to Map Manager QT Mamm Genome 10 327–334 Occurrence Handle10.1007/s003359900997 Occurrence Handle1:CAS:528:DyaK1MXhvFyqtLY%3D Occurrence Handle10087288
PD Markel JC DeFries TE Johnson (1995) ArticleTitleUse of repeated measures in ethanol-induced loss of righting reflex in inbred long-sleep and short-sleep mice Alcohol Clin Exp Res 19 299–304 Occurrence Handle1:CAS:528:DyaK2MXmtFygtrY%3D Occurrence Handle7625561
PD Markel DW Fulker B Bennett RP Corley JC DeFries et al. (1996) ArticleTitleQuantitative trait loci for ethanol sensitivity in the LS × SS recombinant inbred strains: Interval mapping Behav Genet 26 447–458 Occurrence Handle1:STN:280:BymA28jnvFU%3D Occurrence Handle8771905
J Markham E Fifkova A Scheetz (1987) ArticleTitleEffect of chronic ethanol consumption on the fine structure of the dentate gyrus in long-sleep and short-sleep mice Exp Neurol 95 290–302 Occurrence Handle10.1016/0014-4886(87)90139-7 Occurrence Handle1:CAS:528:DyaL2sXhtlemtbo%3D Occurrence Handle3803516
GE McClearn R Kakihana (1981) Selective breeding for ethanol sensitivity: Short-sleep and long-sleep mice GE McClearn RA Deitrich VG Erwin (Eds) Development of Animal Models as Pharmacogenetic Tools U.S. Government Printing Office Washington, DC 81–113
GE McClearn JR Wilson W Meredith (1970) The use of isogenic and heterogenic mouse stocks in behavioral research G Lindzey DD Thiessen (Eds) Contributions to Behavior–Genetic Analysis: The Mouse As a Prototype Appleton-Century-Crofts New York 3–22
J Owens B Bennett TE Johnson (2002) ArticleTitlePossible pleiotropic effects of genes specifying sedative/hypnotic sensitivity to ethanol on other alcohol-related traits Alcohol Clin Exp Res 26 1461–1467 Occurrence Handle1:CAS:528:DC%2BD38XotVCmtrw%3D Occurrence Handle12394278
W Poelchen WR Proctor TV Dunwiddie (2000) ArticleTitleThe in vitro ethanol sensitivity of hippocampal synaptic gamma-aminobutyric acid (a) responses differs in lines of mice and rats genetically selected for behavioral sensitivity or insensitivity to ethanol J Pharmacol Exp 295 741–746 Occurrence Handle1:CAS:528:DC%2BD3cXnsl2kt7c%3D Occurrence Handle11046113
BA Rikke JE Yerg ME Battaglia TR Nagy DB Allison et al. (2003) ArticleTitleStrain variation in the response of body temperature to dietary restriction Mech Ageing Dev 124 663–678 Occurrence Handle10.1016/S0047-6374(03)00003-4 Occurrence Handle12735906
BA Rikke JE Yerg ME Battaglia TR Nagy DB Allison et al. (2004) ArticleTitleQuantitative trait loci specifying the response of body temperature to dietary restriction J Gerontol 59A 11
Stenehjem S, Bruggemann E (2001) Glume bar phenotypes in a b73 × mo17 recombinant inbred population reveal the epistatic interaction between p11 and b1. In: Marize Genetic Conference, 42nd Annual Marize Genetic Conference, Coeur d’Alene, ID (http://www.maizegdb.org/maize-meeting/2001/03abstracts.pdf, p 80)
A Stinchcomb BJ Bowers JM Wehner (1989) ArticleTitleThe effects of ethanol and ro 15-4513 on elevated plus-maze and rotarod performance in long-sleep and short-sleep mice Alcohol 5 369–376 Occurrence Handle10.1016/0741-8329(89)90006-2
KM Swanberg JR Wilson A Kalisker (1979) ArticleTitleDevelopmental and genotypic effects on pituitary–adrenal function and alcohol tolerance in mice Dev Psychobiol 12 201–210 Occurrence Handle1:CAS:528:DyaE1MXktFCqu7o%3D Occurrence Handle437360
BA Taylor (1978) Recombinant inbred strains: Use in gene mapping HC Morse (Eds) Origins of Inbred Mice Academic Press New York 423–435
CM Wade EJ Kulbokas AW Kirby MC Zody JC Mullikin et al. (2002) ArticleTitleThe mosaic structure of variation in the laboratory mouse genome Nature 420 574–578 Occurrence Handle10.1038/nature01252 Occurrence Handle1:CAS:528:DC%2BD38Xpt1WhsL4%3D Occurrence Handle12466852
GS Wand (1989) ArticleTitleEthanol differentially regulates proadrenocorticotropin/endorphin production and corticosterone secretion in LS and SS lines of mice Endocrinology 124 518–526 Occurrence Handle1:CAS:528:DyaL1MXntV2lsg%3D%3D Occurrence Handle2535813
GS Wand (1990) ArticleTitleDifferential regulation of anterior pituitary corticotrope function is observed in vivo but not in vitro in two lines of ethanol-sensitive mice Alcohol Clin Exp Res 14 100–106 Occurrence Handle1:CAS:528:DyaK3cXktVWru74%3D Occurrence Handle1689969
RW Williams J Gu S Qi L Lu (2001) ArticleTitleThe genetic structure of recombinant inbred mice: High-resolution consensus maps for complex trait analysis Genome Biol 2 IssueID11 46–46
Acknowledgments
We thank Jerome Salazar and Jean Wah for careful and reliable animal care and breeding and Eugene Thomas for the initial breeding. We also thank Nancy Phares–Zook for her research contributions and for her outstanding job managing our many diverse databases. Linda Lou Wessman, Justin Springett, and Jessica Hall cut many tails and extracted DNA. Lena Gordon ran numerous screening and confirmatory gels. We also thank our colleagues at IBG and elsewhere who gave advice throughout the study and in reading the many versions of the manuscript. We thank Shuhua Qi for excellent support in genotyping the LXS strains and Yanhua Qu for helping with the BLAT analysis of microsatellite sequences. Dr. Elissa Chesler patiently and on numerous occasions gave BB very thorough instruction on application and interpretation of R/qtl. This work was supported by grants from the NIH [RO1 AA11984-(TEJ) and KO1 AA00195 (TEJ) and P50 AA13755 (RWW)], by the Ellison Foundation for Medical Research (TEJ), and by funds from the University of Colorado. Genotyping costs were supported in part by P20-MH 62009 (a Human Brain Project to RWW) and the JNIA Genotyping Core (U24AA13513 to LL and RWW).
Author information
Authors and Affiliations
Additional information
(Robert W. Williams and Beth Bennett)These authors contributed equally to this work.
Rights and permissions
About this article
Cite this article
Williams, R.W., Bennett, B., Lu, L. et al. Genetic structure of the LXS panel of recombinant inbred mouse strains: a powerful resource for complex trait analysis. Mamm Genome 15, 637–647 (2004). https://doi.org/10.1007/s00335-004-2380-6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00335-004-2380-6