
Abstract
Polar bears in the Beaufort (SB) and Chukchi (CS) Seas experience different environments due primarily to a longer history of sea ice loss in the Beaufort Sea. Ecological differences have been identified as a possible reason for the generally poorer body condition and reproduction of Beaufort polar bears compared to those from the Chukchi, but the influence of exposure to other stressors remains unknown. We use molecular technology, quantitative PCR, to identify gene transcription differences among polar bears from the Beaufort and Chukchi Seas as well as captive healthy polar bears. We identified significant transcriptional differences among a priori groups (i.e., captive bears, SB 2012, SB 2013, CS 2013) for ten of the 14 genes of interest (i.e., CaM, HSP70, CCR3, TGFβ, COX2, THRα, T-bet, Gata3, CD69, and IL17); transcription levels of DRβ, IL1β, AHR, and Mx1 did not differ among groups. Multivariate analysis also demonstrated separation among the groups of polar bears. Specifically, we detected transcript profiles consistent with immune function impairment in polar bears from the Beaufort Sea, when compared with Chukchi and captive polar bears. Although there is no strong indication of differential exposure to contaminants or pathogens between CS and SB bears, there are clearly differences in important transcriptional responses between populations. Further investigation is warranted to refine interpretation of potential effects of described stress-related conditions for the SB population.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Increasingly, gene transcription-based diagnostics of wildlife are being used in the assessment of health, not only in individuals or populations but also in ecosystems (Acevedo-Whitehouse and Duffus 2009). Concurrent with the need to understand cumulative impacts of environmental stressors on wildlife are advances in molecular technology capable of identifying the synergistic effects of multiple stressors on the individual physiology of different species (Bowen et al. 2012, 2015, Miles et al. 2012). Gene transcription analyses can be particularly useful in identifying the mechanisms of subtle but significant deleterious effects of disturbances on wildlife. Gene transcription is physiologically driven by the extent of intrinsic and extrinsic stimuli such as infectious agents, toxins, trauma, or neoplasia. The earliest observable signs of health impairment are altered levels of gene transcripts, evident prior to clinical manifestation (McLoughlin et al. 2006), thus providing an early warning of potentially compromised health and, concurrently, environmental stressors on individuals, populations, and ecosystems. However, perhaps due to the novel nature of this technology, transcript-based technologies have been slow to develop for some sensitive species and ecosystems.
Polar bears (Ursus maritimus) from the Chukchi Sea (CS) and southern Beaufort Sea (SB) have exhibited different responses to recent changing environmental conditions, possibly due in part to existing ecosystem disparities. Given this disparity in population-level responses, these polar bears are ideal candidates for identifying and detecting systemic perturbations with transcript-based tools. Whereas sea ice loss has been attributed to declines in body condition and cub and adult survival in the SB (Regehr et al. 2007; Rode et al. 2010; Bromaghin et al. 2015), CS polar bears have maintained body condition and cub production during a period of substantial sea ice loss (Rode et al. 2014). The CS is characterized by a vast continental shelf where sea depth rarely exceeds 300 m and marine primary productivity is among the highest of the Arctic Ocean (Sakshaug 2004). The CS has experienced a relatively short history of reduced sea ice habitat. By contrast, the SB has a relatively narrow continental shelf which gives way to some of the deepest, and comparatively less productive, waters of the Arctic Ocean (Sigler et al. 2011). Since 1979, the CS and SB have experienced dramatic reductions in the extent of summer sea ice, characterized by lengthening melt seasons (i.e., the period of time between the onset of sea ice melt in the spring and freeze-up in the fall) that have increased at rates of ≈9 days/decade and ≈12 days/decade, respectively (Stroeve et al. 2014). However, due to its narrow continental shelf, the SB experiences an annual open-water period (i.e., substantive lack of sea ice habitat over the shelf) that is about twice as long (94 vs. 44 days) as in the CS (Rode et al. 2014). These ecological differences have been hypothesized as possible reasons for the generally poorer body condition and reproduction of SB polar bears compared to those from the CS (Rode et al. 2014).
Additionally, polar bears can be exposed to high levels of contaminants; levels of many organohalogens were found to be greater in the SB than those in the CS subpopulation, suggesting differences in exposure sources between the two regions (Kannan et al. 2005; Letcher et al. 2010). Lately, it has become clear that climate change has the potential to alter the fate of contaminants in the environment by changing the point sources, transport, distribution, bioavailability, environmental stability, and toxicity/ecotoxicology (e.g., Kallenborn et al. 2012; Moe et al. 2013). For instance, reduced sea ice extent constrains the ability of polar bears to access their primary prey, ringed seals (Pusa hispida) (Derocher et al. 2004; Ferguson et al. 2005), which has the potential to alter trophic interactions and mediate increased body burdens of legacy contaminants as polar bears switch to foraging on alternate prey species. For example, in the western Hudson Bay region of the Canadian Arctic, declining sea ice cover has been accompanied by increases in the proportion of open-water seal species [e.g., harp (Phoca groenlandica) and harbor (Phoca vitulina) seals] represented in polar bear diets, which have been associated with increased concentrations of several chlorinated and brominated contaminants in polar bear adipose tissue (McKinney et al. 2009, 2013). Studies have found associations between high levels of legacy chlorinated contaminants in certain polar bear subpopulations and biomarkers of toxic effects on endocrine, immune, and reproductive functions (Braathen et al. 2004; Lie et al. 2004, 2005; Sonne 2010; Simon et al. 2011; Bechshoft et al. 2012). Consequently, climate change may result in increased stress of toxic compounds to polar bear populations (McKinney et al. 2009; Kallenborn et al. 2012; Moe et al. 2013).
Development of a tool with which to identify physiological changes at the molecular level may help identify specific factors affecting the overall health of polar bears and their corresponding Arctic ecosystem. However, a key requirement for the evaluation of the changing condition of a population or ecosystem is the establishment of reference values with which individuals or populations potentially at risk can be compared (Mancia et al. 2008). Consequently, initial transcript-based analyses of polar bear populations must be made prior to the occurrence of further environmental change.
Herein, we describe the development of species-specific quantitative real-time polymerase chain reaction (qPCR) assays to measure differential transcript levels of 14 genes in the polar bear (Table 1). The genes examined in our study can be grouped into functional categories that include immune modulation, pathogen response, inflammation, cell signaling, xenobiotic-metabolizing enzymes, and cellular stress response, and are fundamental to mediation of detoxification and immune function (Schwartz et al. 2004a, b), cellular injury (Ghanem et al. 2006), signal transduction (Burchiel and Luster 2001), xenobiotic metabolism (Schwartz et al. 2004a, b), or tumorigenesis (Ramesh et al. 2004) (Table 1). The genes targeted in our study were largely selected based upon their potential to be modified by biological, physical, or anthropogenic injury, and may provide information on the type and magnitude of stressors associated with the animal’s internal or external environment. We compared transcription of targeted genes among polar bears sampled in the SB and CS, representing two populations with different types and magnitudes of environmental stressors, and clinically healthy, captive individuals. Our objective was to develop a baseline set of transcript values for three unique populations of polar bears from which comparisons might be made with future samples. Ultimately, these methods may provide an understanding of the susceptibility of individuals and subsequently populations at risk from dynamic regime shifts such as seen with climate change in the Arctic, and provide an early-warning indicator for species at risk.
Materials and methods
Free-ranging target polar bears
Ninety-four SB polar bears were captured in spring (late March–early May) 2012 and 2013, and 50 CS bears were captured in spring 2013. Approximately equal numbers of males and females were captured in each location. Details concerning capture date, location, and sex of the free-ranging bears are found in Table 2. Polar bears were immobilized from a helicopter using Telazol® (tiletamine hydrochloride and zolazepam hydrochloride; Zoetis, Florham Park, NJ)-filled projectile syringes fired from a dart gun. Captured bears were ear-tagged with a unique identification number that was also tattooed on both sides of the inner surface of the upper lip, and age was established using various methods. Cubs-of-the-year were always with their mothers and could be visually aged without error (Ramsay and Stirling 1988). Some bears had been captured and marked in previous years, so their age was determined from their capture history. For bears other than dependent young that had no capture history, we extracted a vestigial premolar and determined age by analysis of cementum annuli (Calvert and Ramsay 1998). Bears were assigned to the following age classes: adult (≥5 yrs), subadult (3–4 yrs), two-year old, yearling, and cubs-of-the-year. A fatness index (FI) was used to determine whether body condition potentially influenced transcription patterns. We determined FI by palpation and assigned individuals a score from 1 (extremely thin) to 5 (obese) based on the distribution of adipose tissue around the body (Stirling et al. 2008). This index correlates positively with lipid concentration of adipose tissue in polar bears (McKinney et al. 2014). FI provides a coarse resolution measure of body condition that is likely less sensitive to changes in body condition compared to continuous metrics such as body mass and length metrics. Details of FI are found in Table 2.
We routinely inspected all individuals and noted the presence and absence of distinguishing marks such as scars, physical abnormalities, and outward symptoms of disease. In 2012, we observed the occurrence of alopecia (i.e., patches of hair loss) on 28 % of animals captured in the southern Beaufort Sea (Atwood et al. 2015). Alopecia is a syndrome characterized by patchy hair thinning and loss and occasional epidermal lesions, often distributed irregularly over the body, and associated with viral, bacterial, fungal, and parasitic agents (e.g., Lynch et al. 2011). For all alopecic bears, we collected tissue samples (e.g., hair plucks, skin biopsies from affected areas, skin scrapes) for pathology. We also collected a subset of the same types of samples from unaffected bears to serve as controls. Details on the alopecia investigation are available in Atwood et al. (2015).
Captive polar bears
Blood samples were obtained from 17 captive, adult, clinically healthy polar bears from Anchorage Zoo (Anchorage, AK), Brookfield Zoo (Brookfield, IL), Buffalo City Zoo (Buffalo, NY), Cleveland Metroparks Zoo (Cleveland, OH), Columbus Zoo (Powell, OH), Detroit Zoo (Royal Oak, MI), Kansas City Zoo (Kansas City, MO), Louisville Zoo (Louisville, KY), Omaha Henry Doorly Zoo (Omaha, NE), Oregon Zoo (Portland, OR), Point Defiance Zoo and Aquarium (Tacoma, WA), SeaWorld Orlando (Orlando, FL), SeaWorld San Diego (San Diego, CA), and the Toledo Zoo (Toledo, OH). Details concerning age and sex of the captive bears are found in Table 3.
Blood collection and RNA extraction
A 2.5-ml sample from each polar bear (captive and free-ranging) was drawn directly into a PAXgene™ blood RNA collection tube (PreAnalytiX, Switzerland) from either the jugular, femoral, or popliteal veins and then frozen at −20 °C until extraction of RNA (Bowen et al. 2012). Rapid RNA degradation and induced transcription of certain genes after blood draws have led to the development of methodologies for preserving the RNA transcription profile immediately after blood is drawn. The PAXgene™ tube contains a blend of RNA stabilizing reagents that protect RNA molecules from degradation by RNases and prevents further induction of gene transcription. Without this stabilization, copy numbers of individual mRNA species in whole blood can change more than 1000-fold during storage and transport. The RNA from blood in PAXgene™ tubes was isolated according to manufacturer’s standard protocols (PreAnalytiX 2009), which included an on-column DNase treatment to remove contaminating gDNA (silica-based microspin technology), and the extracted RNA stored at −80 °C until analysis. All RNA was checked for quality on a nanodrop 2000 and achieved A260/A280 ratios of approximately 2.0 and A260/A230 ratios of less than 1.0.
cDNA synthesis
A standard cDNA synthesis was performed on 2 μg of RNA template from each animal. Reaction conditions included 4 units of reverse transcriptase (Omniscript®, Qiagen, Valencia, CA), 1 μM random hexamers, 0.5 mM each dNTP, and 10 units of RNase inhibitor, in RT buffer (Qiagen, Valencia, CA). Reactions were incubated for 60 min at 37 °C, followed by an enzyme inactivation step of 5 min at 93 °C, and then stored at −20 °C until further analysis.
PCR primers
Degenerate primers were designed from multi-species alignments (GenBank) as previously described (Bowen et al. 2007). Briefly, degenerate primer pairs developed for the polar bear were utilized on cDNA from three randomly selected polar bear samples. Degenerate primer pairs were designed to amplify 14 genes of interest and one ribosomal housekeeping gene (Bowen et al. 2007). PCR amplifications using these primers were performed on 20 ng of each cDNA sample in 50 μl volumes containing 20–60 pmol of each primer, 40 mM Tris-KOH (pH 8.3), 15 mM KOAc, 3.5 mM Mg (OAc)2, 3.75 μg/ml bovine serum albumin (BSA), 0.005 % Tween-20, 0.005 % Nonidet-P40, 200 μM each dNTP, and 5U of Advantage® 2 Taq polymerase (Clontech, Palo Alto, CA). The PCR was performed on an MJ Research PTC-200 thermal cycler (MJ Research, Watertown, MA) and consisted of 1 cycle at 94 °C for 3 min, and then 40 cycles at 94 °C for 30 s, at 60 °C for 30 s, and 72 °C for 2 min, with a final extension step of 72 °C for 10 min. The products of these reactions were electrophoresed on 1.5 % agarose gels and resulting bands visualized by ethidium bromide staining. Definitive bands representing PCR products of a predicted base pair size of the targeted gene were excised from the gel, and extracted and purified using a commercially available nucleic acid-binding resin (Qiaex II Gel extraction kit, Qiagen, Valencia, CA).
Nucleotide sequences of isolated fragments were determined by dideoxy nucleotide methodology using an automated sequencer (Model 373; Applied Biosystems, Foster City, CA). Nucleotide sequences of the PCR products were analyzed using Align™ and Contig™ sequence alignment software programs (Vector NTI™; InforMax Inc., North Bethesda, MD) and compared to known sequences using the NCBI BLAST program (Altschul et al. 1990) and the IMGT/HLA database (Robinson et al. 2001). Primer pairs appropriate for real-time PCR were designed based on the elucidated polar bear sequences for each gene (Table 4).
Real-time PCR
Real-time PCR systems for the individual, polar bear-specific reference, or housekeeping gene (S9) and genes of interest were run in separate wells (Table 1). Briefly, 1 µl of cDNA was added to a mix containing 12.5 μl of QuantiTect Fast SYBR Green® Master Mix [5 mM Mg2+] (Qiagen, Valencia, CA), 0.5 μl each of forward and reverse sequence-specific primers, and 10.5 μl of RNase-free water; total reaction mixture was 25 μl. The reaction mixture cDNA samples for each gene of interest and the S9 gene were loaded into MicroAmp Fast Optical® 96-well reaction plates in duplicate and sealed with optical sealing tape (Applied Biosystems, Foster City, CA). Reaction mixtures containing water, but no cDNA, were used as negative controls; thus, approximately two individual polar bear samples were run per plate.
Amplifications were conducted on a Step One Plus Real-time Thermal Cycler (Applied Biosystems, Foster City, CA) using Step One Software v. 2.2.2. Reaction conditions were as follows: 95 °C for 20 s, 40 cycles of 95 °C for 3 s and 60 °C for 30 s. The melt curve consists of 95 °C for 15 s, 60 °C for 1 min, 0.3 °C per second temperature increase, and then 95 °C for 15 s. Reaction specificity was monitored by melting curve analysis using a final data acquisition phase during a step and hold process of a 0.3 °C per second temperature increase from 60 to 95 °C. Cycle threshold crossing values (C T) for the genes of interest were normalized to the S9 housekeeping gene.
Statistical analysis
Analysis of qPCR data was conducted using normalized values, i.e., housekeeping gene threshold crossing (in qPCR, the point at which amplification is exponential) subtracted from the gene of interest threshold crossing for each animal (McLoughlin et al. 2006).
We used nonparametric statistical analyses because the cycle threshold (C T) measure of gene transcription provided by qPCR may have a lognormal distribution (McLoughlin et al. 2006). We tested the assumption that transcript levels of our genes of interest were lognormally distributed. Geometric means and 95 % upper and lower confidence limits were computed, and Anderson–Darling and Shapiro–Wilk tests for normality were performed for all genes; we used two tests as a conservative measure of normality, and Spearman’s correlation matrix values were analyzed for all genes to identify relationships among genes (NCSS, Statistical and Power Analysis Software, 2007, Kaysville, UT, USA). First, we used two-way analysis of similarities (ANOSIM, Primer E) to examine potential interaction between sex and age on transcript values. We then used conventional mean responses per classification group (Captive bears, Beaufort 2012, Beaufort 2013, Chukchi 2013) with data assessed for statistical significance between classification ranks using Kruskal–Wallis with Dunn’s Multiple Comparison Tests to determine relative transcription levels among groups (NCSS).
We used nonparametric statistical analyses because the cycle threshold (C T) measure of gene transcription provided by qPCR may have a lognormal distribution (McLoughlin et al. 2006). We used ANOSIM (R Core Team 2013) analysis of variance to test for differences: in gene transcription among groups, i.e., the SB 2012, SB 2013, CS 2013, and CAP. We used discriminant function analysis to identify genes contributing to separation of groups (NCSS, Statistical and Power Analysis Software, 2007, Kaysville, UT, USA). The genes identified by discriminant function analysis as significant were then subjected to multivariate, nonmetric, multi-dimensional scaling analysis (NMDS; R Core Team 2013) in conjunction with cluster analysis for statistical and graphical representation of individual polar bears clustered by similarity in transcription and not by predefined groups such as location. Statistical significance was based on p values ≤0.05, and in the case of the ANOSIM tests, relative to the R Statistic value.
We used analysis of variance (ANOVA) to compare mean FI values of free-ranging adult polar bears between subpopulations (SB and CS), between years, and within reproductive classes (for adult females, i.e., solitary female or female with dependent young) (Table 2). Polar bear body condition can change rapidly, so we included ordinal date of capture as a covariate in all analyses (Rode et al. 2014). Because we had multi-year data for the SB polar bears, we ran multiple sets of analyses on these data, including separate analyses for each year. The 2012 SB polar bear data included 10 adults (7 males, 3 females) that were affected by the alopecia syndrome, which was associated with reduced body condition (Atwood et al. 2015). For adult females, main effects included year, ordinal date of capture, and reproductive class. For adult males, year and ordinal date of capture were included as covariates. We then compared FI scores of adult females and males captured in 2013 between subpopulations using the same sets of covariates with the exception of year. Statistical significance was based on p values ≤0.05.
Results
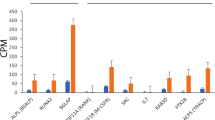
Only four genes (AHR, IL1β, T-bet, and Gata3) had transcript values following normal distributions. All genes had unimodal distributions; deviations from normal involved right (HSP70) or left (COX2, DRβ) skewness. Variation (based on confidence intervals) was small for most genes except HSP70 (Table 5). Correlation matrices identified several significant strong correlations between genes, i.e., Gata3 and THRα (r = 0.77, p = 0.00), TGF-beta (or TGFβ) and THRα (r = 0.75, p = 0.00), AHR and THRα (r = 0.72, p = 0.00), Gata3 and TGFβ (r = 0.71, p = 0.00), Gata3 and T-bet (r = 0.67, p = 0.00), and T-bet and THRα (r = 0.67, p = 0.00). No differences were found for any genes between age groups, but differences between sexes were identified for THRα (p = 0.01), TGFβ (p = 0.02), and Gata3 (p = 0.04), which all had a higher transcription in females compared to males. No interactions between sex and age were identified (p > 0.05).
We identified significant transcriptional differences among a priori groups (i.e., captive bears, SB 2012, SB 2013, CS 2013) for ten of the 14 genes of interest (i.e., CaM, HSP70, CCR3, TGFb, COX2, THRa, T-bet, Gata3, CD69, and IL17); transcription levels of DRβ, IL1β, AHR, and Mx1 did not differ among groups (Table 5). Discriminant function analysis of transcription response revealed eight genes (CaM p = 0.00; CCR3 p = 0.00; COX2 p = 0.04; Gata3 p = 0.02; HSP70 p = 0.00; T-bet p = 0.00; TGFβ p = 0.00; THRα p = 0.02) that contributed significantly to the separation of groups. Overall, gene transcription (C T) values differed among the four polar bear groups (ANOSIM, p < 0.001, Global R = 0.12).
When analyzed without a priori structure (e.g., group), polar bears separated into fairly well-defined groups as depicted by NMDS (Fig. 1). Refinement of the ordination from two- to three-dimensional space improved the adequacy of the NMDS analysis (i.e., stress improved from 0.15 to 0.07, whereas 0.05 is defined as excellent representation), and the clusters were visually separated in three-dimensional space.

Multivariate, nonparametric, multi-dimensional scaling analysis of transcription response of 14 targeted genes in polar bears sampled in the Beaufort Sea 2012 (BS12) and 2013 (BS13), Chukchi Sea 2013 (CH13), and captives (CAP). Three-dimensional stress = 0.07. Little overlap exists among clusters
Fatness index (FI) values of free-ranging bears were similar between years, subpopulations, and female reproductive class. For adult females in the SB, FI values varied by year (F 1,36 = 5.20, p = 0.03), with values in 2012 (i.e., the year of the aforementioned alopecia syndrome) lower than in 2013 regardless of reproductive status (F 1,36 = 2.51, p = 0.12) or ordinal date (F 1,36 = 0.18, p = 0.67). Fatness index values for adult females captured in the SB and CS in 2013 did not differ between subpopulation, ordinal date of capture, and reproductive status (F 3,34 = 0.36, p = 0.78). For adult males in the SB, FI values did not differ between years or by date of capture (F 2,34 = 1.02, p = 0.37), nor did FI values differ between males captured in the SB and CS in 2013 (F 2,39 = 0.35, p = 0.71).
Discussion
We identified significant transcriptional differences among a priori groups for ten of the 14 genes of interest. Additionally, fatness index values for adult females in the SB varied by year, with values in 2012 lower than in 2013 regardless of reproductive status or date of capture. Although distinct transcript patterns may exist, it can be difficult to disentangle the effects of environmental factors on these pathways, as (1) pathways are interconnected and (2) stressors may impinge on several of the pathways simultaneously (Kennerly et al. 2008). In cases where a relatively small gene panel is used, it may be more effective to examine responses on a per gene basis.
Our initial intent was to use the clinically healthy captive polar bears as a control or baseline population. However, there are stresses and conditions associated with captivity which preclude the use of captives as a reference range; there is no population free of all external or internal stimuli. Although determined to be “healthy,” these captive polar bears were likely experiencing at least some degree of physiologic perturbation, albeit at a level consistent with subclinical manifestation. Thus, in the absence of longitudinal data, the determination of a baseline is unlikely and comparisons must be relative, with the findings in one individual or population being compared and contrasted with other individuals and populations. It is important to note in this case that increased levels of transcription among groups or individuals may be perfectly normal responses to stimuli and are likely an indication of a properly functioning system.
For example, up-regulation of AHR is indicative of immediate exposure to classes of environmental toxicants including polycyclic aromatic hydrocarbons, polyhalogenated hydrocarbons, dibenzofurans, and dioxin (Oesch-Bartlomowicz and Oesch 2005). Chronic exposure to specific toxicants may not necessarily cause a sustained increase in AHR transcription (Bowen et al. 2007), but can be associated with potential downstream consequences, e.g., modulation of T-regulatory (T REG) (immune-suppressive) or T-helper type 17 (T H17) (pro-inflammatory) immunologic activity (Quintana et al. 2008; Veldhoen et al. 2008). IL17 transcription was higher in SB than in CS polar bears possibly reflecting the history of oil and gas development along the coast of the SB, even though transcription of AHR was not different between SB and CS polar bears. Although the SB polar bears probably are in closer proximity to the existing oil and gas activity, we cannot give a firm conclusion on whether the higher IL17 transcription in the SB population could be associated with emission from petroleum activities or is due to other ecological or environmental differences between the two populations.
Excessive production of TH17 cells has been suggested as key in the development of autoimmunity, which can result in symptoms such as dermatitis and other skin disorders (Parham 2014). Consistent with results from human studies, stress responses, evidenced from elevated TGFβ levels, were also documented in bycatch harbor porpoises (Phocoena phocoena; Fonfara et al. 2007). In humans, elevated TGFβ has been shown to induce TH17 production, while CD69 suppresses it (Martin and Sanchez-Madrid 2011; Parham 2014). In general, CD69 transcript levels were lower in BS 2012 than in all other groups, while transcript levels of TGFβ were higher in both SB groups than in CS or captive polar bears. Further, COX2 transcription can also be an indicator of stress as is the case in many captive wild animals (McPhee and Carlstead 2010; Bowen et al. 2012); levels of COX2 were higher in both captive and Beaufort Sea polar bears than in Chukchi bears.
In 2012, some of the SB polar bears sampled for this study were observed with varying degrees of alopecia (Atwood et al. 2015), as well as reduced body condition (via analysis of FI values) for adult females captured during this period. Polar bears were not captured in the CS in 2012, and it is unknown whether the CS population was similarly affected by alopecia in 2012. Concurrent with the observation of alopecia in polar bears, elevated numbers of alopecic ringed seals were detected in the Beaufort, Chukchi, and Bering seas in 2011 and 2012, resulting in the declaration of an unusual mortality event by the National Oceanic and Atmospheric Administration (NOAA 2011). The agents causing these cases of alopecia are unknown, and related physiological changes within individual animals have been undetectable using classical diagnostic methods such as serology, blood chemistries, toxicology screenings, and cytology of lesion biopsies. However, the transcriptional patterns we observed suggest differences in biological processes associated with immune response, viral defense, and stress response between individuals with and without alopecia; genes such AHR, HSP70, IL1β, CD69, DRB, MX1, and TGFβ were differentially transcribed. The higher transcription of select genes and finding of reduced body condition in 2012 adult SB females may be related. SB polar bears may experience more stressful conditions, i.e., longer history of melting sea ice, industrial development, and lower primary productivity, than CS polar bears.
A positive correlation was found between transcription of Gata3 and T-bet overall. From classical human immunology, negative correlation between these two genes is expected, as they are part of a negative feedback loop regulating “opposing” branches of the immune system (Parham 2014). However, interpretations derived largely from studies on transcription in humans should be undertaken with caution.
Accordingly, further investigation of a potential linkage among transcription patterns, immune function, and physical condition is warranted. Examination of transcriptional responses of additional genes that indicate organic exposure and nutritional stress is also warranted, particularly given the differences observed between CS and SB polar bears. While both the CS and the SB have experienced dramatic declines in the extent of summer sea ice habitat, body size, condition, and recruitment indices are higher for CS bears, indicating that the CS population appears to be in better condition than SB bears (Rode et al. 2014). These key differences may be mediated by geographic variation in prey availability, exposure to contaminants, pathogens, or both. Although melting sea ice may release globally transported contaminants that distill into polar regions and sequester in ice (Pfirman et al. 1995, 1997), there is no strong indication of differential exposure to contaminants or pathogens between CS and SB bears. By contrast, there is evidence that SB polar bears are nutritionally stressed when compared to CS bears: Cherry et al. (2009) estimated that ≈22 % of adult females sampled in the Beaufort Sea during the spring of 2005 and 2006 were fasting, whereas Rode et al. (2014) found no evidence of fasting adult females sampled in the CS during 2008–2011. Increased contaminant burdens in marine mammals can alter the expression of genes associated with thyroid function, xenobiotic metabolism, and immunological response (Gregory and Cyr 2003; Buckman et al. 2011), and fasting has been shown to augment those disruptive effects (e.g., Vijayan et al. 2006). Linking genes associated with contaminant exposure and nutritional stress to those examined in our current study would refine interpretation of potential effects of described stress-related conditions for the SB population.
Our results represent a foundation upon which gene transcription panels can contribute to comprehensive analyses that provide better interpretation of polar bear population health and response to changing environmental conditions, i.e., as effects of climate change become more pronounced. Such an approach will enhance the sensitivity and value of the polar bear as a sentinel species for marine ecosystem health and provide a valuable tool for monitoring the cumulative impacts of biotic and abiotic stressors on vulnerable polar bear subpopulations (Vongraven et al. 2012).
References
Acevedo-Whitehouse K, Duffus ALJ (2009) Effects of environmental change on wildlife health. Phil Trans R Soc Lond B 364:3429–3438
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Atwood TC, Peacock E, Burek-Huntington K, Shearn-Bochsler V, Bodenstein B, Durner G, Beckmann K (2015) Prevalence and spatio-temporal variation of an alopecia syndrome detected in polar bears in the southern Beaufort Sea. J Wildl Dis 51:48–59. doi:10.7589/2013-11-301
Bechshoft TO, Sonne C, Dietz R, Born EW, Muir DCG, Letcher RJ, Novak MA, Henchey E, Meyer JS, Jenssen BM, Villanger GD (2012) Associations between complex OHC mixtures and thyroid and cortisol hormone levels in East Greenland polar bears. Environ Res 116:26–35
Bowen L, Aldridge B, Miles AK, Stott JL (2006) Expressed MHC class II genes in sea otters (Enhydra lutris) from geographically disparate populations. Tissue Agn 67:402–408
Bowen L, Schwartz J, Aldridge B, Riva F, Miles AK, Mohr FC, Stott JL (2007) Differential gene expression induced by exposure of captive mink to fuel oil: a model for the sea otter. EcoHealth 4:298–309
Bowen L, Miles AK, Murray M, Haulena M, Tuttle J, Van Bonn W, Adams L, Bodkin JL, Ballachey BE, Estes JA, Tinker MT, Keister R, Stott JL (2012) Gene transcription in sea otters (Enhydra lutris); emerging diagnostics in marine mammal and ecosystem health. Mol Ecol Resour 12:67–74. doi:10.1111/j.1755-0998.2011.03060.x
Bowen L, Miles AK, Kolden CA, Saarinen JA, Bodkin JL, Murray MJ, Tinker MT (2015) Effects of wildfire on sea otter (Enhydra lutris) gene transcription profiles. Mar Mamm Sci 31:191–210. doi:10.1111/mms.12151
Braathen M, Derocher AE, Wiig Ø, Sørmo EG, Lie E, Skaare JU, Jenssen BM (2004) Relationships between PCBs and thyroid hormones and retinol in female and male polar bears. Environ Health Perspect 112:826–833
Bromaghin JF, McDonald TL, Stirling I, Derocher AE, Richardson ES, Regehr EV, Douglas DC, Durner GM, Atwood TC, Amstrup SC (2015) Polar bear population dynamics in the southern Beaufort Sea during a period of sea ice decline. Ecol Appl 25:634–651
Buckman AH, Veldhoen N, Ellis G, Ford JK, Helbing CC, Ross PS (2011) PCB-associated changes in mRNA expression in killer whales (Orcinus orca) from the NE Pacific Ocean. Enviro Sci Technol 45:10194–10202
Burchiel SW, Luster MI (2001) Signaling by environmental polycyclic aromatic hydrocarbons in human lymphocytes. Clin Immunol 98:2–10
Calvert W, Ramsay MA (1998) Evaluation of age determination of polar bears by counts of cementum growth layer groups. Ursus 10:449–453
Chen Z, Wang H, Matsumura K, Qian P (2012) Expression of calmodulin and myosin light chain kinase during larval settlement of the barnacle Balanus Amphitrite. PLoS ONE 7:e31337
Cherry SG, Derocher AE, Stirling I, Richardson ES (2009) Fasting physiology of polar bears in relation to environmental change and breeding behavior in the Beaufort Sea. Polar Biol 32:383–391
De Maio A (1999) Heat shock proteins: facts, thoughts, and dreams. Shock 11:1–12
Derocher AE, Lunn NJ, Stirling I (2004) Polar bears in a warming climate. Integr Comp Biol 44:163–176
Dong L, Ma Q, Whitlock JP Jr (1997) Down-regulation of major histocompatibility complex Q1b gene expression by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. J Biol Chem 272:29614–29619
Ferguson SH, Stirling I, McLoughlin P (2005) Climate change and ringed seal (Phoca hispida) recruitment in western Hudson Bay. Mar Mamm Sci 21:121–135
Fonfara S, Siebert U, Prange A, Colijn F (2007) The impact of stress on cytokine and haptoglobin mRNA expression in blood samples from harbour porpoises (Phocoena phocoena ). J Mar Biol Assoc UK 87:305–311
Ghanem MM, Batelli LA, Mercer RR, Scabilloni JF, Kashon ML, Ma JY, Nath J, Hubbs AF (2006) Apoptosis and Bax expression are increased by coal dust in the polycyclic aromatic hydrocarbon-exposed lung. Environ Health Perspect 114:1367–1373
Goldsby RA, Kindt TJ, Osborne BA, Kuby J (2003) Immunology, 5th edn. WH Freeman and Company, New York
Gregory M, Cyr DG (2003) Effects of environmental contaminants on the endocrine system of marine mammals. In: Vos JG, Ross PS, de Swart RL, van Loveren H, Osterhaus AD (eds) Toxicology of marine mammals, volume 3—systems. Taylor and Francis, New York, pp 67–81
Gurish MF, Humbles A, Tao H, Finkelstein S, Boyce JA, Gerard C, Friend DS, Austen KF (2002) CCR3 is required for tissue eosinophilia and larval cytotoxicity after infection with Trichinella spiralis. J Immunol 168:5730–5736
Harris SG, Padilla J, Koumas L, Ray D, Phipps RP (2002) Prostaglandins as modulators of immunity. Trends Immunol 23:144–150
Iwama GK, Mathilakath MV, Forsyth RB, Ackerman PA (1999) Heat shock proteins and physiological stress in fish. Am Zool 39:901–909
Kallenborn R, Halsall C, Dellong M, Carlsson P (2012) The influence of climate change on the global distribution and fate processes of anthropogenic persistent organic pollutants. J Environ Monit 14:2854–2869
Kannan K, Yun SH, Evans TJ (2005) Chlorinated, brominated, and perfluorinated contaminants in livers of polar bears from Alaska. Environ Sci Technol 39:9057–9063
Kennerly E, Ballmann A, Martin S, Wolfinger R, Gregory S, Stoskopf M, Gibson G (2008) A gene expression signature of confinement in peripheral blood of red wolves (Canis rufus). Mol Ecol 17:2782–2791. doi:10.1111/j.1365294X.2008.03775.x
Kibenge MJT, Munir K, Kibenge FSB (2005) Constitutive expression of Atlantic salmon Mx1 protein in CHSE-214 cells confers resistance to infectious salmon Anaemia virus. Virol J 2:75
Kringel H, Iburg T, Dawson H, Aasted B, Roepstorff A (2006) A time course study of immunological responses in Trichuris suis infected pigs demonstrates induction of a local type 2 response associated with worm burden. Int J Parasitol 36:915–924
Letcher RJ, Bustnes JO, Dietz R, Jenssen BM, Jorgensen EH, Sonne C, Verreault J, Vijayan MM, Gabrielsen GW (2010) Exposure and effects assessment of persistent organohalogen contaminants in arctic wildlife and fish. Sci Total Environ 408:2995–3043
Lie E, Larsen HJS, Larsen S, Johansen GM, Derocher AE, Lunn NJ, Norstrom RJ, Wiig Ø, Skaare JU (2004) Does high organochlorine (OC) exposure impair the resistance to infection in polar bears (Ursus maritimus)? Part I: effect of OCs on the humoral immunity. J Toxicol Environ Health A 67:555–582
Lie E, Larsen HJS, Larsen S, Johansen GM, Derocher AE, Lunn NJ, Norstrom RJ, Wiig Ø, Skaare JU (2005) Does high organochlorine (OC) exposure impair the resistance to infection in polar bears (Ursus maritimus)? Part II: possible effects of OCs on mitogen- and antigen-induced lymphocyte proliferation. J Toxicol Environ Health A 68:457–484
Lynch M, Kirkwood R, Mitchell A, Duignan P, Arnould JPY (2011) Prevalence and significance of an alopecia syndrome in Australian fur seals (Arctocephalus pusillus doriferus). J Mamm 92:342–351
Martin P, Sanchez-Madrid F (2011) CD69: an unexpected regulator of TH17 cell-driven inflammatory responses. Sci Signal 4:pe14. doi:10.1126/scisignal.2001825
McKinney MA, Peacock E, Letcher RJ (2009) Sea ice-associated diet change increases the levels of chlorinated and brominated contaminants in polar bears. Environ Sci Technol 43:4334–4336
McKinney MA, Iverson SJ, Fisk AT, Sonne C, Riget FF, Letcher RJ, Arts MT, Born EW, Rosing-Asvid A, Dietz R (2013) Global Change effects on the long-term feeding ecology and contaminant exposures of East Greenland polar bears. Glob Chang Biol 19:2360–2372
McKinney MA, Atwood T, Dietz R, Sonne C, Iverson SJ, Peacock E (2014) Validation of adipose lipid content as a body condition index for polar bears. Ecol Evol 4:516–527
McLoughlin K, Turteltaub K, Bankaitis-Davis D, Gerren R, Siconolfi L, Storm K, Cheronis J, Trollinger D, Macejak D, Tryon V, Bevilacqua M (2006) Limited dynamic range of immune response gene expression observed in healthy blood donors using RT-PCR. Mol Med 12:185–195
McPhee ME, Carlstead K (2010) The importance of maintaining natural behaviors in captive mammals. In: Kleiman DG, Thompson KV, Baer CK (eds) Wild mammal in captivity: principles and techniques for zoo management. University of Chicago Press, Illinois, pp 303–313
Miles AK, Bowen L, Ballachey B, Bodkin JL, Murray M, Keister RA, Stott JL (2012) Variations of transcript profiles between sea otters (Enhydra lutris) from Prince William Sound, Alaska, and clinically normal reference otters. Mar Ecol Prog Ser 451:201–212
Moe SJ, De Schamphelaere K, Clements WH, Sorensen MT, Van den Brink PJ, Liess M (2013) Combined and interactive effects of global climate change and toxicants on populations and communities. Environ Toxicol Chem 32:49–61
National Oceanic and Atmospheric Administration (NOAA) (2011) 2011 Arctic seal disease outbreak update on investigation and findings. Alaska, U.S. Department of Commerce http://alaskafisheries.noaa.gov/protectedresources/seals/ice/diseased/factsheet112211
Oesch-Bartlomowicz B, Oesch F (2005) Phosphorylation of cytochromes P450: first discovery of a posttranslational modification of a drug-metabolizing enzyme. Biochem Biophys Resour Commun 338:446–449
Parham P (2014) The Immune System, 4th edn. Garland Science, Taylor and Francis Group, LLC, New York
Pfirman S, Eicken H, Bauch D, Weeks WF (1995) The potential transport of pollutants by Arctic sea ice. Sci Total Environ 159:129–146
Pfirman S, Kogeler JW, Rigor I (1997) Potential for rapid transport of contaminants from the Kara Sea. Sci Total Environ 202:111–122
PreAnalytiX (2009) PAXgene blood RNA Kit handbook, 2nd edn. QIAGEN GmbH for PreAnalytix, Switzerland, pp 37–39
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Ramesh A, Walker SA, Hood DB, Guillen MD, Schneider K, Weyand EH (2004) Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol 23:301–333
Ramsay MA, Stirling I (1988) Reproductive biology and ecology of female polar bears (Ursus maritimus). J Zool 214:601–634
Regehr EV, Lunn NJ, Amstrup SC, Stirling I (2007) Effects of earlier sea ice breakup on survival and population size of polar bears in Western Hudson Bay. J Wildl Manag 71:2673–2683
Robinson J, Waller M, Parham P, Bodmer JG, Marsh SGE (2001) IMGT/HLA Database—a sequence database for the human major histocompatibility complex. Nucl Acids Res 29:210–213
Rode KD, Amstrup SC, Regehr EV (2010) Reduced body size and cub recruitment in polar bears associated with sea ice decline. Ecol Appl 20:768–782
Rode KD, Regehr EV, Douglas D, Durner G, Derocher AE, Thiemann G, Budge S (2014) Variation in the nutritional and reproductive ecology of two polar bear populations experiencing sea ice loss. Glob Change Biol 20:76–88. doi:10.1111/gcb.12339
Sakshaug E (2004) Primary and secondary production in the Arctic Seas. In: Stein R, MacDonald RW (eds) The organic carbon cycle in the Arctic Ocean. Springer, Berlin, pp 57–81
Schwartz JA, Aldridge BM, Lasley BL, Snyder PW, Stott JL, Mohr FC (2004a) Chronic fuel oil toxicity in American mink (Mustela vison): systemic and hematological effects of ingestion of a low-concentration of bunker C fuel oil. Toxi Appl Pharm 200:146–158
Schwartz JA, Aldridge BM, Lasley BL, Snyder PW, Stott JL, Mohr FC (2004b) Immunophenotypic and functional effects of bunker C fuel oil on the immune system of American mink (Mustela vison). Vet Immunol Immunopathol 101:179–190
Sigler MF, Renner M, Danielson SL, Eisner LB, Lauth RR, Kuletz KJ, Logerwell EA, Hunt GL Jr (2011) Fluxes, fins, and feathers: relationships among the Bering, Chukchi, and Beaufort seas in a time of climate change. Oceanography 24:250–265
Simon E, Bytingsvik J, Jonker W, Leonards PEG, de Boer J, Jenssen BM, Lie E, Aars J, Hamers T, Lamoree MH (2011) Blood plasma sample preparation method for the assessment of thyroid hormone-disrupting potency in effect-directed analysis. Environ Sci Technol 45:7936–7944
Sonne C (2010) Health effects from long-range transported contaminants in Arctic top predators: an integrated review based on studies of polar bears and relevant model species. Environ Int 36:461–491
Stirling I, Thiemann GW, Richardson E (2008) Quantitative support for a subjective fatness index for immobilized polar bears. J Wildl Manag 72:568–574
Stroeve JC, Markus T, Boisvert L, Miller J, Barrett A (2014) Changes in Arctic melt seasons and implications for sea ice loss. Geophys Res Lett 41:1216–1225. doi:10.1002/2013GL058951
Tabuchi M, Veldhoen N, Dangerfield N, Jeffries S, Helbing CC, Ross PS (2006) PCB-related alteration of thyroid hormones and thyroid hormone receptor gene expression in free-ranging harbor seals (Phoca vitulina). Environ Health Perspect 114:1024–1031
Tsai MJ, O’Malley BW (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63:451–486
Tsan MF, Gao B (2004) Cytokine function of heat shock proteins. Am J Physiol-Cell Physiol 286:C739–C744
Tumpey TM, Szretter KJ, Hoeven NV, Katz JM, Kochs G, Haller O, García-Sastre A, Staeheli P (2007) The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol 81:10818–10821
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109
Vijayan MM, Aluru N, Maule AG, Jørgensen EH (2006) Fasting augments PCB impact on liver metabolism in anadromous arctic char. Tox Sci 91:431–439
Vongraven D, Aars J, Amstrup SC, Atkinson SN, Belikov S, Born EW, DeBruyn T, Derocher AE, Gill M, Lunn NJ, Obbard ME, Omelak J, Ovsyanikov NG, Peacock E, Richardson E, Sahanatien V, Stirling I, Wiig Ø (2012) A circumpolar monitoring framework for polar bears. Ursus 23:1–66
Wegner KM, Kalbe M, Rauch G, Kurtz J, Schaschl H, Reusch TBH (2006) Genetic variation in MHC class II expression and interactions with MHC sequence polymorphism in three-spined sticklebacks. Mol Ecol 15:1153–1164
Acknowledgments
Funding was provided by the U.S. Geological Survey’s Changing Arctic Ecosystems Initiative. We are grateful for the contributions of the veterinarians to the Anchorage Zoo, SeaWorld San Diego, Brookfield Zoo, Cleveland Metroparks Zoo, SeaWorld Orlando, Louisville Zoo, Detroit Zoo, Kansas City Zoo, Omaha Henry Doorly Zoo, Buffalo City Zoo, Toledo Zoo, Columbus Zoo, Oregon Zoo, and Point Defiance Zoo and Aquarium. Samples were provided by the zoos and aquaria under the expressed permission of the US Fish and Wildlife Service, and under the Animal Care Committee protocols established at each of the aquaria. Mention of trade names or organizations does not imply endorsement by the US government.
Conflict of interest
We are not aware of any conflicts of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bowen, L., Miles, A.K., Waters, S. et al. Gene transcription in polar bears (Ursus maritimus) from disparate populations. Polar Biol 38, 1413–1427 (2015). https://doi.org/10.1007/s00300-015-1705-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-015-1705-0