
Abstract
Phylogenetic diversity of the marine bacterioplankton in Kongsfjorden (Spitsbergen) was investigated by 16S rRNA gene analysis. Community fingerprint analysis by PCR-denaturing gradient gel electrophoresis revealed that there was no apparent difference of bacterioplankton community composition between sampling locations in the fjord. A higher biodiversity was observed in bottom water of station 3 in the central part of the fjord. By 16S rRNA gene clone library analysis, sequences detected both in surface and bottom water of station 3 fell into eight putative divisions, including Proteobacteria (Alpha, Beta, Gamma and Delta), Bacteroidetes, Actinobacteria, Verrucomicrobia and unidentified bacteria, in addition to chloroplasts of algae. Sequences representing Planctomycetes were only observed in bottom water. Compared to the preponderance of clones representing Gammaproteobacteria (36.5%) and Alphaproteobacteria (29.4%) in bottom water, Alphaproteobacteria (43.6%) and algae (27.7%) constituted two dominant fractions in surface water. Cloned sequences showed 82.1–100% similarity to those described sequences. It suggests that, attributing to the influence of ocean currents as well as freshwater input in the summer, the bacterial community in Kongsfjorden may consist of a mixture of cosmopolitan and uniquely endemic phylotypes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Kongsfjorden, located on the west coast of the Spitsbergen (Svalbard) archipelago at 79°N, is a glacial and open fjord in the Arctic. The water column in Kongsfjorden is strongly influenced both by Atlantic and Arctic water masses (Hop et al. 2002). At the same time, tidal glaciers at the head of the fjord discharge freshwater and suspended loads that cause steep environmental gradients in salinity, temperature and sedimentation rates along the length of this fjord, as well as affecting bottom sediment composition, and thus conditions change from a stable marine system at the fjord entrance to a very unstable brackish system in the inner fjord basin (Weslawski et al. 2000). Due to its location as a border area between the Atlantic and Arctic biogeographical zones, Kongsfjorden has received much research attention in the recent past. The current interest in the fjord is primarily based on the fact that Kongsfjorden is highly suitable as a site for exploring the impacts of possible climate change, with both Atlantic water influx and melting of tidal glaciers being linked to climate variability (Hop et al. 2002).
Marine diversity is currently one of the most studied topics in ecology especially under the framework of global and regional environmental changes. Habitat and environmental heterogeneity generally favor biodiversity (Laudien et al. 2007). Being major components of food webs, bacterioplankton play key roles in biogeochemical cycles and energy flow in marine ecosystems, especially in the assimilation and mineralization of dissolved organic carbon in the ocean carbon cycle (Mou et al. 2008). In Kongsfjorden, the average bacterial abundance in the outer and inner part is 2.29 × 108 and 3.52 × 108 cells l−1, respectively, and the mean biomass distribution for all investigated stations is 82.58% for bacteria (Jiang et al. 2005). However, to date there is little information concerning the phylogenetic composition of the bacterioplankton community in this fjord. A better understanding of the bacterial community composition would yield insight into the potential impact of global climate changes on the marine biodiversity in this ecosystem.
Recently developed techniques of molecular biology permit microbiologists to estimate microbial community composition and diversity in complex habitats. With the introduction of these methods, it has become possible to carry out detailed evaluations of the biodiversity of aquatic microbial communities via 16S rRNA gene cloning and sequencing (Dorigo et al. 2005). Several recent studies have used these techniques to analyze the composition and diversity of marine bacteria in Svalbard. In cold fjords of Svalbard the bacterial isolates associated with sea ice fall into five phylogenetic groups, which are the alpha and gamma subclasses of Proteobacteria, low and high G+C Gram positives and the Cytophaga–Flavobacterium–Bacteroides (CFB) group (Groudieva et al. 2004). The phylogenetic composition of the bacterial community in sediments includes Beta-, Gamma- and Deltaproteobacteria, CFB and Planctomycetales (Ravenschlag et al. 2001). Strains isolated from the seasonal snow pack in Spitsbergen around Ny-Alesund belong to the Alpha-, Beta- and Gammaproteobacteria, Firmicutes and Actinobacteria (Amato et al. 2007).
Using a combination of denaturing gradient gel electrophoresis (DGGE), amplified ribosomal DNA restriction analysis (ARDRA) and 16S rRNA gene clone library, the aim of the present work was to investigate the community composition of bacterioplankton in Kongsfjorden by analysis of its 16S rRNA gene sequences. This is a beginning toward an understanding of the planktonic microbial communities in Kongsfjorden, Spitsbergen.
Materials and methods
Sample collection and DNA extraction
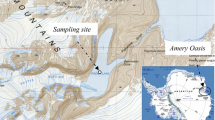
Water samples were collected at the surface and bottom depths during the Chinese Arctic Yellow River Station Expedition 2007 (10 July to 10 August 2007) to investigate the phylogenetic composition of the bacterioplankton community in Kongsfjorden, Spitsbergen. Station locations are shown in Fig. 1. Table 1 gives the characteristics of water samples for bacterial community analysis. Surface water samples were collected by a bucket, while bottom water samples were collected with Niskin bottles; 1.5–2 l of water was pressure-filtered through 0.2-μm pore size polycarbonate membrane filters (Whatman, UK). The filters were placed in sterile 2-ml centrifuge tubes and covered with 1.5 ml lysis buffer (Bosshard et al. 2000). After processing, the tubes were immediately frozen and stored at −20°C until used. Total community DNA extraction was carried out as described by Bosshard et al. (2000) and Bano and Hollibaugh (2000). The molecular weight and concentration of DNA in extracts were determined by ethidium bromide-UV detection on 1% (w/v) agarose gel. Nucleic acid extracts from each sample were also analyzed spectrophotometrically at 260 and 280 nm using a DU800 spectrophotometer (Beckman Coulter, Fullerton, CA, USA).

Location of sampling stations in Kongsfjorden, Spitsbergen
PCR–DGGE analysis
Variation in bacterial community composition was assessed by PCR–DGGE. The v3 region of the eubacterial 16S rRNA gene was amplified using forward primer 341f with GC clamp (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG CAG CAG-3′) and a universal reverse primer 534r (5′-ATT ACC GCG GCT GCT GG-3′) (Li et al. 2006; Muyzer et al. 1993). At least 1 ng of genomic DNA was used as a template in a 50 μl PCR reaction mixture containing 50 μM dNTPs, 0.2 μM of each described primers, 5 μl of 10× PCR buffer and 1 U Taq DNA polymerase (Takara, Dalian, China). PCR amplification was performed under the following conditions: initial denaturation of the template DNA at 94°C for 5 min, a touchdown PCR using ten cycles consisting of denaturation at 94°C for 1 min, annealing at 65°C (the temperature was decreased by 1°C every second cycle until the touchdown temperature of 56°C was reached) for 1 min and primer extension at 72°C for 45 s, and then 20 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 45 s, with a final extension at 72°C for 8 min. DGGE was performed using a DCode™ Universal Mutation Detection System (Bio-Rad, Hercules, CA, USA). For each sample, 400 ng of PCR product of v3 region was loaded on an 8% (w/v) polyacrylamide gel with a gradient of denaturant between 40 and 60%. A denaturing gradient consisting of 100% denaturant is defined as 7 M urea with 40% (v/v) formamide. Electrophoresis was carried out at 170 V for 5 h (with an initial 30 min at 40 V) at 60°C in 1× TAE buffer. Gels were stained with ethidium bromide for 30 min and visualized under UV irradiance. Gel images were captured with a Gel Doc XR documentation system. DGGE bands migrating to the same position in different lanes in the gel were considered to have the same sequence. The banding patterns of DGGE profiles were analyzed by Quantity One 4.6 software (Bio-Rad, Hercules, CA, USA).
16S rRNA gene PCR
The extracted DNA from the seawater was suitable for direct use as a PCR template. Almost full-length 16S rRNA gene was amplified from genomic DNA by PCR with a universal bacterial primer pair 8f (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 1492r (5′-GGT TAC CTT GTT ACG ACT T-3′) (Bosshard et al. 2000). Amplification was carried out in 50-μl reactions with an Eppendorf Mastercycler Gradient (Eppendorf, Hamburg, Germany) as described by Zeng et al. (2007). The success of PCRs was determined by electrophoresis of 4 μl of the reaction mixture in 1% (w/v) agarose gel in 0.5× TAE buffer with a 2,000-bp DNA ladder (Takara, Dalian, China) as a size marker. Gels were stained with ethidium bromide and examined using a Gel Doc XR documentation system (Bio-Rad, Hercules, CA, USA).
Clone library construction
The 16S rRNA gene-PCR products of the total community were ligated to the vector pMD-18T (Takara, Dalian, China), and used to transform Escherichia coli DH5α competent host cells. The transformed cells were plated on Luria–Bertani (LB) plates containing 100 μg ml−1 of ampicillin. About 150 colonies were randomly selected from the plates for each library and grown overnight in LB broth containing 100 μg ml−1 of ampicillin. Clone inserts were PCR amplified with vector-specific standard M13 primers (M13forGT: 5′-TGT AAA ACG ACG GCC AGT and M13rev-C: 5′-CAG GAA ACA GCT ATG ACC) to determine the presence and size of the inserts. For the PCR, 3 μl of each clone culture was used directly as a DNA template in a 25-μl PCR reaction mixture containing 100 μM dNTPs, 0.4 μM of each described primers, 2.5 μl of 10× PCR buffer and 1 U Taq DNA polymerase. The PCR reaction started with pre-denaturation at 95°C for 8 min followed by 25 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 68°C for 1 min, with a final extension at 68°C for 10 min. PCR products were determined using 1% (w/v) agarose gel electrophoresis. Clones that produced a band with an approximate size of 1,500 bp, which corresponded with the expected size of the cloned 16S rRNA gene fragment, were chosen for ARDRA.
ARDRA of a clone library
For screening the clone library for unique types, ARDRA of the PCR products was performed. Amplification products of target size were digested with 10 U of the restriction endonuclease AluI (Takara, Dalian, China) for 4 h at 37°C. The resulting bands were then analyzed by 2% (w/v) agarose gel electrophoresis (Zeng et al. 2007). Clones with identical ARDRA pattern were considered members of the same operational taxonomy unit (OTU). At least one clone of each representative OTU was sequenced.
Sequencing of 16S rRNA gene PCR fragments
A selected DGGE band, representative of bands with the same mobility in the gel, was excised from the gel. Excised band was re-amplified and cloned as described by Li et al. (2005). Clone was checked on the DGGE to ensure the correct mobility. The cloned DGGE bands, as well as clones of each representative OTU described above, were sequenced using M13 primers with an ABI Prism 3730 DNA analyzer (PE Applied Biosystems, Foster City, CA, USA).
Phylogenetic analysis
The 16S rRNA gene sequences obtained were checked for chimeras with Chimera Check from the ribosomal database project (RDP) II (Maidak et al. 2001), and compared to those in GenBank using BLAST search (http://www.ncbi.nlm.nih.gov) and to those in the RDP using SEQUENCE MATCH tool (http://rdp.cme.msu.edu). The closest matches were aligned together with the clone sequences using CLUSTALX 1.81 program (Thompson et al. 1997). A phylogenetic tree was inferred using the PHYLIP v.3.5c software package (Felsenstein 1993). Evolutionary distance matrices, generated by DNADIST, were constructed following the method of Kimura (1980). The matrices were used to infer dendrograms using the neighbor-joining method (Saitou and Nei 1987). Tree figures were generated using NJplot version 2.1 (Perrière and Gouy 1996).
Nucleotide sequence accession number
Sequences reported in this study have been deposited in the GenBank database and assigned GenBank accession numbers: EU919753–EU919862.
Results
Bacterial community composition as assessed by PCR–DGGE
As shown in Fig. 2a, most DGGE bands were shared by five water samples, indicating the similar bacterial community composition of the surface seawater from the outer to the inner part of Kongsfjorden. However, some DGGE bands, for example, represented by bands s10 and s13, showed a difference in band strength between sampling locations (higher in stations 1 and 5).

Denaturing gradient gel electrophoresis fingerprints of bacterial communities in water samples collected in Kongsfjorden, western Spitsbergen. Bands indicated by a small letter and number were excised for sequencing. a Surface water, b bottom water
The bacterial community profiles of the five investigated sites were compared by cluster analysis. The resulting dendrogram of DGGE patterns from surface water samples showed two clusters (Fig. 3a). Community composition of the inner part (station 5) was more similar to the outer part (station 1) than to the middle region (stations 2, 3 and 4) of the fjord.

Cluster analysis of DGGE patterns of bacterial communities in water samples. a Surface water, b bottom water
As shown in Fig. 2b, DGGE profiles of bottom water showed that many bands, represented by bands from b1 to b11 as well as bands from b17 to b21, were common to the five samples, indicating the similar community composition of bacteria in bottom water from the outer to the inner part of Kongsfjorden. At the same time, compared to the outer parts of stations 1 and 2, samples from the central (station 3) and inner parts (stations 4 and 5) of Kongsfjorden contained additional bands, such as bands b14 and b15, suggesting a relatively higher diversity of bacteria in these locations.
The dendrogram of DGGE patterns from bottom water samples (Fig. 3b) showed that bacterial communities of stations 1 and 2 formed a cluster separated from the cluster consisting of stations 3, 4 and 5.
Phylogenetic analysis of DGGE bands
A total of 39 individual bands, consisting of 18 from surface water and 21 from bottom water, were obtained from the DGGE gel (Fig. 2). The lengths of the resolved partial 16S rRNA gene sequences varied from 164 to 192 nucleotides. Table 2 presents the phylogenetic affiliations and similarity values of the most closely related GenBank sequences for all sequences of DGGE bands obtained in this study. DGGE bands were similar (92.2–100%) to their closely related GenBank sequences that all originated from uncultured organisms and most (82%) from aquatic environments, while 15% were related to Arctic bacteria.
Sequences of DGGE bands from surface water fell into seven bacterial phylogenetic clusters: Alpha-, Gamma- and Epsilonproteobacteria, Bacteroidetes, Actinobacteria, Firmicutes and cyanobacteria. Based on the number of DGGE bands, Alphaproteobacteria (22.2% of total bands), Gammaproteobacteria (22.2%) and Bacteroidetes (22.2%) were the three major bacterial components in surface water.
It is worth mentioning that the surface water contained chloroplasts, including members of the Prasinophycean and Ostreococcus (Table 2). In addition, sequences of bands s14 and s18 showed 98 and 99% similarity to two Roseobacter bacteria (Table 2), respectively.
Sequences of DGGE bands from bottom water fell into six major bacterial divisions: Alpha-, Beta- and Gammaproteobacteria, Bacteroidetes, Actinobacteria and Verrucomicrobia. Alphaproteobacteria (33.3%), Gammaproteobacteria (19.0%) and Bacteroidetes (28.5%) were also the three major bacterial components in bottom water. However, sequences related to phototrophic microorganisms, including cyanobacteria and algae, were not found in bottom water.
The community composition of bacteria in the sea water at station 3, which showed the possibility of higher biodiversity based on DGGE fingerprints (Fig. 2), were further investigated using 16S rRNA gene clone library analysis.
16S rRNA gene clone library
Two 16S rRNA gene clone libraries were constructed from environmental DNA of surface and bottom water from station 3. The randomly selected 151 and 150 recombinants from surface and bottom water resulted in 94 and 126 clones showing a single band of approximately 1,500 bp, respectively.
ARDRA of the clone library
Figure 4 shows an example of the ARDRA patterns obtained. Each ARDRA pattern corresponded to one OTU, or a particular phylotype. Analysis of AluI restriction patterns of 16S rRNA gene allowed the grouping of 94 and 126 clones from the surface and bottom water, respectively, into 41 and 76 different OTUs. Most of ARDRA patterns were composed by only one or two clones, suggesting a high degree of interspecific genetic diversity (Michaud et al. 2004). Values of the Simpson’s diversity index of clone library from surface and bottom water were 0.899 and 0.971, respectively, suggesting a higher diversity in bottom water than that in surface water. In addition, there were seven ARDRA patterns shared by the two clone libraries, indicating that seven bacterial phylotypes were distributed both in surface and bottom water at station 3.

Example of ARDRA patterns of amplified 16S rRNA gene digested with endonuclease AluI. M DNA marker DL2000 (Takara, Dalian, China)
Phylogenetic analysis of the clone library
Representative clones showing unique ARDRA patterns were sequenced. The inserted 16S rRNA gene sequences from the clones were aligned to the most similar ones available in the GenBank database. Results showed that the cloned sequences were similar (with similarity values ranging from 82.1 to 100%) to their closely related GenBank sequences, >91% originating from uncultured organisms and most from aquatic environments. The percentage of cloned sequences for which the highest similarity was below 98% and below 93% was 12.7 and 3.2%, respectively.
Coverage (C) was used to quantify how much the environmental diversity was described by a clone library. It was estimated by the equation C = [1−(n 1/N)] × 100%, where N is the number of clones being examined and n 1 represents the number of clone types occurring only once (Giovannoni et al. 1995). The estimation of coverage was 71.3 and 61.9% for surface and bottom water, respectively, indicating that most of the bacteria groups could be detected from the 16S rRNA gene clone library.
As shown in Fig. 5, consistent with DGGE profiles, 16S rRNA gene clone library from surface water showed that Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes and algae were the four major components of the microbial community of surface water in Kongsfjorden.

Clone library composition for surface and bottom water collected from station 3 in Kongsfjorden. Prokaryotes were displayed at the division level (and also subdivision level for Proteobacteria)
Compared to DGGE profiles, a higher microbial diversity was observed in bottom water by 16S rRNA gene clone library analysis (Fig. 5). Gamma- and Alphaproteobacteria constituted the two dominant fractions in bottom water, consistent with the results of DGGE profiles. However, Bacteroidetes only occupied a small fraction of the local community in bottom water shown by clone library analysis.
Similar sequences were detected between DGGE and clone library analysis: in surface water, sequences of bands s7 and s12 showed 100% similarity to clone s111 within the Gammaproteobacteria, and clone s32 within the algae, respectively. Sequence of band b10 from bottom water shared 100% similarity to clone b149 within the Actinobacteria.
All seven clones clustered within the Planctomycetes and only detected in bottom water were very similar (98.3–99.5%) to reported sequences from marine environments, including Pacific Ocean water and deep-sea octacoral (data from GenBank). Two cloned sequences, represented by clone b47 (EU919776), showed 99.3% similarity to uncultured Blastopirellula sp. S25_997 (EF574653). However, sequence specific for anammox Planctomycetes (Schmid et al. 2000) was not detected in the seven clones.
The phylogenetic positions of clones belonging to different OTUs are shown in Fig. 6. Clones s1 (82.1% similarity) and s10 (83% similarity) from surface water, and clone b40 (88.2% similarity) from bottom water formed a separate cluster of unidentified bacteria in phylogenetic trees, indicating the existence of novel microbial species.

Phylogenetic trees showing the affiliation of 16S rRNA gene clones with selected sequences in GenBank database. a Surface water, b bottom water. The total number of clones showing the same sequence is given in square brackets. Asterisk in the phylogenetic trees indicates sequence found both in surface and bottom water. The scale bar indicates evolutionary distance, as calculated using Kimura’s two-parameter distance calculation (substitutions per nucleotide)
A total of 16 clones (17% of the total clones) in clone library of surface water, represented by clone s32 (EU919837), showed 98% similarity to alga Mantoniella squamata (X90641). Chloroplasts from algae possessed 27.7% of the clones from surface water, while the proportional value decreased to 0.8% in bottom water (Fig. 5). This result is consistent with DGGE profiles indicating that algae live in surface water and are not in bottom water.
In clone library of surface water (Fig. 6a), a total of 23 clones (24.5%), represented by clone s3 (EU919824) affiliated with the order Rhizobiales in the Alphaproteobacteria, showed the same sequence. They were closely related (99.8% similarity) to one uncultured bacterial clone 2C228566 (EU800458) from Delaware Bay in America (Shaw et al. 2008). In clone library of bottom water (Fig. 6b), 14 clones (11.1%), represented by clone b23, had the same sequence with clone s3, indicating that this Rhizobiales species was abundant both in surface and bottom water.
Two cloned sequences within the family Oceanospirillaceae in the Gammaproteobacteria, represented by clone s28 (EU919834) from surface water, were 100% identical to seven cloned sequences (represented by clone b39) from bottom water (Fig. 6), indicating that some bacterial species were distributed throughout the whole water column. Sequences of these clones were closely related (99.7% similarity) to one uncultured marine bacterium Ant4D3 (DQ295237) from Antarctic seawater (Grzymski et al. 2006). Represented by clone b60 (EU919782) from bottom water, another seven clones with the same sequence affiliated with the Gammaproteobacteria, showed 99.9% similarity to an uncultured organism clone ctg_NISAA05 (DQ396099) from deep-sea octacoral. These two Gammaproteobacteria species were relatively abundant in bottom water.
By using RDP Classifier to identify the cloned sequences, a greater microbial diversity of the different microbial taxa at the genus level was found in bottom water than that in surface water (Fig. 6). For instance, compared to one genus Methylophilus in the Betaproteobacteria detected in surface water clone library, additional four genera Aquabacterium, Curvibacter, Limnobacter and Thiobacillus within the same class were found in bottom water clone library.
At the same time, difference in bacterial taxa at the genus level was observed between the surface and bottom water (Fig. 6). For instance, genus Desulfocapsa was detected in surface water, while genus Nitrospina within the same class Deltaproteobacteria was found in bottom water.
Discussion
As an open fjord, Kongsfjorden is influenced both by Atlantic and Arctic waters, as well as the glacial melt in summer time (MacLachlan et al. 2007). Compared to the values of bottom water (in the range 34.5–34.9), salinity values of surface water in the five investigated locations varied from 30.0 to 31.7 (Table 1), indicating an apparent influence by fresh water in upper water layers. Wind and fresh water are the most important factors influencing water masses in the upper water column layer (Harmes et al. 2007). The zooplankton community has been shown to be largely influenced by advection (Basedow et al. 2004). Similar bacterial community composition in surface water from the outer to the inner part of the fjord was observed by PCR–DGGE. At the same time, difference in DGGE band strength between sampling locations was found. For the limited sensitivity of detection of rare community members by DGGE (Vallaeys et al. 1997), the difference in DGGE profiles of surface water in this fjord may be attributed to the variation of species abundance, which contributes to the template concentration for PCR and further affects the DGGE analysis (Delong and Pace 2001).
Different from surface water, the influence of freshwater on bottom water tended to decrease with the increasing of depth from the inner (station 5; 30 m) to the outer part (station 1; 200 m) in the fjord (Table 1). Though similar bacterial community composition in bottom water from the outer to the inner part of the fjord was also observed (Fig. 2b), bacterial communities of the outer part (stations 1 and 2 at depth 200 m) formed a cluster separately from the cluster of inner (station 4 at depth 75 m and station 5 at depth 30 m) and central (station 3 at depth 200 m) parts by cluster analysis (Fig. 3b). It suggests that, for bottom water, station 3 in the central part of the fjord is a remote area affected by freshwater input in the summer.
Showing close phylogenetic relationship, DGGE bands s8 and s12 were 96 and 99% similar, respectively, to chloroplast sequences of Ostreococcus tauri strain RCC116 and uncultured Prasinophycean EBAC36H07, both from marine environments (Table 2). Represented by clones s29 (EU919835) from surface water and b78 (EU919793) from bottom water, a total of 27 clones showed 89–99% similarity to chloroplast sequences of phototrophic eukaryotes from different sea areas, including the Arabian Sea, Biscay Bay, Cape Hatteras and Liaodong Gulf (data from GenBank), suggesting that algae detected in Kongsfjorden originated from marine environments.
Phototrophic microorganisms usually distribute in upper water layers and have higher abundance in summer (Murphy and Haugen 1985; Keck et al. 1999; Hop et al. 2002). In this study, chloroplasts from algae possessed 27.7% of the clones from surface water, while the proportional value decreased to 0.8% in bottom water. This result was consistent with the chlorophyll a concentration in waters: chlorophyll a concentration in surface water was 0.67 μg l−1 (Table 1), while the value decreased to 0.01 μg l−1 at a depth of 44 m.
Represented by band s18 and clone s9 (EU919826), sequences within the Roseobacter clade were detected in surface water both by DGGE and clone library with similarity values >99%. However, sequence affiliated with the Roseobacter clade was not found in bottom water. Members of the Roseobacter lineage have the trait of aerobic anoxygenic photosynthesis, oxidize carbon monoxide, and produce the climate-relevant gas dimethylsulfide through the degradation of algal osmolytes (Wagner-Döbler and Biebl 2006). It suggests that the distribution of Roseobacter bacteria in surface water has a relationship with the algae.
Alphaproteobacteria, Gammaproteobacteria and Bacteroidetes were the three major bacterial populations both in surface and bottom water in Kongsfjorden, accounting for 63.8 and 71.5% of clones from surface and bottom water, respectively. Similar dominant bacterial populations are also reported in previous studies of the Arctic Ocean or North Sea: Alphaproteobacteria, Gammaproteobacteria and CFB account for 77% of the clones from Central Arctic Ocean samples (Bano and Hollibaugh 2002). At the same time, sequences related to Alphaproteobacteria are abundant (52%) in the mixed layer, while sequences related to Gammaproteobacteria are more abundant (44%) in halocline samples (Bano and Hollibaugh 2002). In this study, Alphaproteobacteria were abundant (43.6%) in surface water, while Gammaproteobacteria were more abundant (36.5%) in bottom water at 200-m depth. In the North Sea, Alphaproteobacteria, Gammaproteobacteria and CFB account for 72% of 410 isolates (Uphoff et al. 2001). However, over 70% of isolates from fjords of Spitsbergen were affiliated with the Gammaproteobacteria (Groudieva et al. 2004), but this may be attributed to the limitation of culture techniques.
Betaproteobacteria usually constitute a dominant fraction in freshwater systems (Glöckner et al. 1999). Sequences of clones b37 (EU919774), b52 (EU919778), b54 (EU919779) and b91 (EU919798) shared 99.7, 99.5, 98.4 and 99.9% similarity, respectively, to uncultured bacterial clone BG.c1 (DQ228368) from Bench Glacier, uncultured bacterial clone 5C231166 (EU803577) from Lake Gatun, uncultured beta proteobacterial clone ADK-SGh02-35 (EF520485) from Adirondack Lake, and uncultured bacterial clone D8A_5 (AY768825) from a deep terrestrial fracture system. On the other hand, close relationships were also found between our clones and sequences from a number of marine environments: sequences of clones s71 (EU919843), b4 (EU919755) and b32 (EU919771) showed significant (99.7, 99.9 and 99.8%) similarity, respectively, to uncultured organism clone ctg_NISA163 (DQ396081) from deep-sea octacoral, uncultured bacterial clone B78-37 (EU287001) from Pacific arctic surface sediment, and uncultured bacterial clone S25_1562 (EF575218) from Coco’s Island. With a higher proportion (7.1%) in bottom water, Betaproteobacteria were detected both in surface and bottom water in Kongsfjorden (Fig. 5). It suggests that the bacterioplankton community in Kongsfjorden is influenced by ocean currents as well as freshwater input in summer time.
Represented by clones b132 (EU919813) and b10 (EU919757), a total of six clones from bottom water showed 98.7–99.7% similarity to sequences of Deltaproteobacteria from marine environments (data from GenBank). At the same time, the sequence of clone s21 (EU919830) from surface water showed 96.5% similarity to an uncultured Desulfocapsa sp. WM24 (DQ133914) from the Frasassi cave system in Italy (Macalady et al. 2006), indicating a novel bacterial species originating from the terrestrial environment. A higher proportion of Deltaproteobacteria, including orders Desulfobacterales (clone b10) and Desulfuromonales (clone b132), was detected in bottom water (4.7%) than that in surface water (1.1%). Including the sulfur-reducing bacteria, the Deltaproteobacteria are a group of chemotrophic bacteria, which constitute an important fraction in bacterial communities of marine sediments, from hydrothermal sediments in the Guaymas Basin to permanently cold marine sediments in Spitsbergen (Ravenschlag et al. 1999; Teske et al. 2002). This suggests that the existence of Deltaproteobacteria in the bottom water of Kongsfjorden may be related to the sulfur cycle in natural anaerobic waters.
In surface water, sequence of DGGE band s16 within the Gammaproteobacteria showed 95% similarity to a sulfur-oxidizing bacterium NDII1.2 (Table 2). At the same time, represented by DGGE bands s4 and b17, sequences affiliated with the Sulfitobacter group in the Alphaproteobacteria were detected both in surface and bottom water. However, no cloned sequence affiliated with the Sulfitobacter group was detected in bottom water by clone library analysis, although a total of six clones in surface water clone library, represented by clone s13 (EU919828), were closely related to Sulfitobacter bacteria. Members of the genus Sulfitobacter specialize on sulfite oxidation (Park et al. 2007). It suggests that the marine bacteria participating in the sulfur cycle by oxidation usually distribute in upper water layers in Kongsfjorden.
With similarity values ranging from 94.3 to 99.5% to the sequences of several other clones obtained from a number of aquatic environments, including sea ice-melt pond, saline lake and deep sea (data from GenBank), sequences affiliated with the Actinobacteria were detected both in surface and bottom water in Kongsfjorden (Fig. 5). Members of the Verrucomicrobia phylum were also detected both in surface and bottom water in Kongsfjorden. Cloned sequences showed 92.2–99.2% similarity to reported sequences from marine environments. It suggests that the Actinobacteria and Verrucomicrobia in this fjord originated from marine environments.
Cloned sequences that clustered with different taxa, including the Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes and Actinobacteria were found to be closely related to previous reported sequences originating from Antarctic aquatic environments. Three cloned sequences (s9, b8 and b94) belonging to the Alphaproteobacteria were very similar (99.6%) to one uncultured marine bacterial clone AntCL1E1 (DQ906726) from Antarctic sea water near Anvers Island. Clustered with the Bacteroidetes, sequence of clone b56 shared 99.1% similarity to uncultured marine bacterial clone AntCL1F9 (DQ906733) from the same Antarctic area. Represented by clone s5, nine cloned sequences clustered with the Gammaproteobacteria were 99.7% similar to uncultured marine bacterium Ant4D3 (DQ295237) from Antarctic coastal water. In addition, sequence of clone s121, clustered with the Actinobacteria, showed 99.3% similarity to uncultured bacterial clone ELB16-004 (DQ015796) from the Lake Bonney, Antarctica. These results suggest the bipolar distribution of cosmopolitan bacteria at the species level. However, analysis at the conservative gene level of 16S rRNA was not sufficient to determine if the same species occurred at both poles, and other analytical methods, including DNA–DNA hybridization and gyrB gene sequencing, are required to elucidate diversity that is not detectable by 16S rRNA gene sequencing (Brinkmeyer et al. 2003).
Compared to DGGE profiles, additional Betaproteobacteria, Deltaproteobacteria and Verrucomicrobia were observed in surface water by 16S rRNA gene clone library analysis, though Epsilonproteobacteria, Firmicutes and cyanobacteria were absent. In bottom water, a higher microbial diversity was detected by 16S rRNA gene clone library analysis. The differences in bacterial community composition detected by DGGE and 16S rRNA gene library reflect the strengths and limitations of the two methods. It suggests that a strategy presented in this study, combining DGGE analysis with the construction of clone library, is an attractive method for investigation of marine bacterial communities.
Cloned sequences showed 82.1–100% similarity to those described sequences, and the percentage of cloned sequences with similarity below 98 was 12.7% in this study. According to a criterion that higher than 93 and 98% similarities correspond to a taxonomic grouping at the genus and species levels, respectively (Mullins et al. 1995; Devereux et al. 1990), it indicates that, attributing to the influence of ocean currents as well as freshwater input in the summer, the bacterial community in Kongsfjorden may consist of a mixture of cosmopolitan and uniquely endemic phylotypes.
In summary, attempts have been made in this study to investigate the community composition of bacterioplankton in arctic glacial Kongsfjorden. No significant variation in bacterial community composition was observed between sampling locations on the basis of PCR-DGGE. By 16S rRNA gene clone library analysis, sequences related to Proteobacteria (Alpha, Beta, Gamma and Delta), Bacteroidetes, Actinobacteria and Verrucomicrobia, as well as chloroplasts from algae, were detected both in surface and bottom water of station 3 in the central part of the fjord, although sequences representing Planctomycetes were only detected in bottom water. Clones with the same 16S rRNA gene sequence were observed both in surface and bottom water, suggesting that some bacterial species were distributed throughout the whole water column. At the same time, there is also a possibility that some bacterial DNA has been sampled in bottom water actually originated from dead cells which sank to the bottom. Gammaproteobacteria and Alphaproteobacteria dominated the planktonic bacteria in bottom water, while Alphaproteobacteria (including Roseobacter group) and algae constituted two dominant fractions in surface water, indicating that phototrophic microorganisms were an important composition in surface water of the fjord during the summer time.
References
Amato P, Hennebelle R, Magand O, Sancelme M, Delort AM, Barbante C, Boutron C, Ferrari C (2007) Bacterial characterization of the snow cover at Spitzberg, Svalbard. FEMS Microbiol Ecol 59:255–264
Bano N, Hollibaugh JT (2000) Diversity and distribution of DNA sequences with affinity to ammonia-oxidizing bacteria of the β subdivision of the class Proteobacteria in the Arctic Ocean. Appl Environ Microbiol 66:1960–1969
Bano N, Hollibaugh JT (2002) Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol 68:505–518
Basedow SL, Eiane K, Tverberg V, Spindler M (2004) Advection of zooplankton in an Arctic fjord (Kongsfjorden, Svalbard). Estuar Coast Shelf Sci 60:113–124
Bosshard PP, Santini Y, Grüter DG, Stettler R, Bachofen R (2000) Bacterial diversity and community composition in the chemocline of the meromictic alpine Lake Cadagno as revealed by 16S rRNA gene analysis. FEMS Microbiol Ecol 31:173–182
Brinkmeyer R, Knittel K, Jürgens J, Weyland H, Amann R, Helmke E (2003) Diversity and structure of bacterial communities in Arctic versus Antarctic pack ice. Appl Environ Microbiol 69:6610–6619
Delong EE, Pace NR (2001) Environmental diversity of bacteria and archaea. Syst Biol 50:470–478
Devereux R, He SH, Doyle CL, Orkland S, Stahl DA, LeGall J, Whitman WB (1990) Diversity and origin of Desulfovibrio species: phylogenetic definition of a family. J Bacteriol 172:3609–3619
Dorigo U, Volatier L, Humbert JF (2005) Molecular approaches to the assessment of biodiversity in aquatic microbial communities. Water Res 39:2207–2218
Felsenstein J (1993) PHYLIP (phylogeny inference package) version 3.5c. Department of Genetics, University of Washington, Seattle
Giovannoni SJ, Mullins TD, Field KG (1995) Microbial diversity in oceanic systems: rRNA approaches to the study of unculturable microbes. In: Joint I (ed) Molecular ecology of aquatic microbes. Springer, Berlin, pp 217–248
Glöckner FO, Fuchs BM, Amann R (1999) Bacterioplankton compositions of lakes and oceans: a first comparison based on fluorescence in situ hybridization. Appl Environ Microbiol 65:3721–3726
Groudieva T, Kambourova M, Yusef H, Royter M, Grote R, Trinks H, Antranikian G (2004) Diversity and cold-active hydrolytic enzymes of culturable bacteria associated with Arctic sea ice, Spitzbergen. Extremophiles 8:475–488
Grzymski JJ, Carter BJ, DeLong EF, Feldman RA, Ghadiri A, Murray AE (2006) Comparative genomics of DNA fragments from six Antarctic marine planktonic bacteria. Appl Environ Microbiol 72:1532–1541
Harmes AAP, Tverberg V, Svendsen H (2007) Physical qualification and quantification of the water masses in the Kongsfjorden–Krossfjorden system cross section. Oceans’07 IEEE, Aberdeen
Hop H, Pearson T, Hegseth EN, Kovacs KM, Wiencke C, Kwasniewski S, Eiane K, Mehlum F, Gulliksen B, Wlodarska-Kowalczuk M, Lydersen C, Weslawski JM, Cochrane S, Gabrielsen GW, Leakey RJG, Lønne OJ, Zajaczkowski M, Falk-Petersen S, Kendall M, Wängberg S-Å, Bischof K, Voronkov AY, Kovaltchouk NA, Wiktor J, Poltermann M, di Prisco G, Papucci C, Gerland S (2002) The marine ecosystem of Kongsfjorden, Svalbard. Polar Res 21:167–208
Jiang X, He J, Cai M (2005) Abundance and biomass of heterotrophic microbes in the Kongsfjorden, Svalbard. Acta Oceanol Sin 24:143–152
Keck A, Wiktor J, Hapter R, Nilsen R (1999) Phytoplankton assemblages related to physical gradients in an arctic, glacier-fed fjord in summer. ICES J Mar Sci 56:203–214
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Laudien J, Herrmann M, Arntz WE (2007) Soft bottom species richness and diversity as a function of depth and iceberg scour in Arctic glacial Kongsfjorden (Svalbard). Polar Biol 30:1035–1046
Li H, Yu Y, Chen B, Zeng Y, Ren D (2005) Molecular genetic diversity of bacteria in the bottom section of arctic sea ice from the Canada Basin. Acta Oceanol Sin 24:153–161
Li H, Yu Y, Chen B, Zeng Y, Ren D (2006) Phylogenetic analysis of bacteria in sea ice brine sampled from the Canada Basin, Arctic Ocean. Chin J Polar Sci 17:81–89
Macalady JL, Lyon EH, Koffman B, Albertson LK, Meyer K, Galdenzi S, Mariani S (2006) Dominant microbial populations in limestone-corroding stream biofilms, Frasassi cave system, Italy. Appl Environ Microbiol 72:5596–5609
MacLachlan SE, Cottier FR, Austin WE, Howe JA (2007) The salinity: δ18O water relationship in Kongsfjorden, western Spitsbergen. Polar Res 26:160–167
Maidak BL, Cole JR, Lilburn TG, Parker CT, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM (2001) The RDP-II (ribosomal database project). Nucleic Acids Res 29:173–174
Michaud L, Di Cello F, Brilli M, Fani R, Lo Giudice A, Bruni V (2004) Biodiversity of cultivable psychrotrophic marine bacteria isolated from Terra Nova Bay (Ross Sea, Antarctica). FEMS Microbiol Lett 230:63–71
Mou X, Sun S, Edwards RA, Hodson RE, Moran MA (2008) Bacterial carbon processing by generalist species in the coastal ocean. Nature 451:708–711
Mullins TD, Britschgi TB, Krest RL, Giovannoni SJ (1995) Genetic comparisons reveal the same unknown bacterial lineages in Atlantic and Pacific bacterioplankton communities. Limnol Oceanogr 40:148–158
Murphy LS, Haugen EM (1985) The distribution and abundance of phototrophic ultraplankton in the North Atlantic. Limnol Oceanogr 30:47–58
Muyzer G, Dewaal EC, Uitterlinden AG (1993) Profiling of complex microbial population by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Park JR, Bae JW, Nam YD, Chang HW, Kwon HY, Quan ZX, Park YH (2007) Sulfitobacter litoralis sp. nov., a marine bacterium isolated from the East Sea, Korea. Int J Syst Evol Microbiol 57:692–695
Perrière G, Gouy M (1996) WWW-Query: an on-line retrieval system for biological sequence banks. Biochimie 78:364–369
Ravenschlag K, Sahm K, Pernthaler J, Amann R (1999) High bacterial diversity in permanently cold marine sediments. Appl Environ Microbiol 65:3982–3989
Ravenschlag K, Sahm K, Amann R (2001) Quantitative molecular analysis of the microbial community in marine arctic sediments (Svalbard). Appl Environ Microbiol 67:387–395
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schmid M, Twachtmann U, Klein M, Strous M, Juretschko S, Jetten M, Metzger JW, Schleifer KH, Wagner M (2000) Molecular evidence for genus level diversity of bacteria capable of catalyzing anaerobic ammonium oxidation. Syst Appl Microbiol 23:93–106
Shaw AK, Halpern AL, Beeson K, Tran B, Venter JC, Martinv JB (2008) It’s all relative: ranking the diversity of aquatic bacterial communities. Environ Microbiol 10:2200–2210
Teske A, Hinrichs KU, Edgcomb V, de Vera Gomez A, Kysela D, Sylva SP, Sogin ML, Jannasch HW (2002) Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl Environ Microbiol 68:1994–2007
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Uphoff HU, Felske A, Fehr W, Wagner-Döbler I (2001) The microbial diversity in picoplankton enrichment cultures: a molecular screening of marine isolates. FEMS Microbiol Ecol 35:249–258
Vallaeys T, Topp E, Muyzer G, Macheret V, Laguerre G, Rigaud A, Soulas G (1997) Evaluation of denaturing gradient gel electrophoresis in the detection of 16S rRNA gene sequence variation in rhizobia and methanotrophs. FEMS Microbiol Ecol 24:279–285
Wagner-Döbler I, Biebl H (2006) Environmental biology of the marine Roseobacter lineage. Annu Rev Microbiol 60:255–280
Weslawski JM, Pedersen G, Falk-Petersen S, Porazinski K (2000) Entrapment of macroplankton in an Arctic fjord, basin, Kongsfjorden, Svalbard. Oceanologia 42:57–69
Zeng Y, Liu W, Li H, Yu Y, Chen B (2007) Effect of restriction endonucleases on assessment of biodiversity of cultivable polar marine planktonic bacteria by amplified ribosomal DNA restriction analysis. Extremophiles 11:685–692
Acknowledgments
We appreciate the assistance of the Chinese Arctic and Antarctic Administration (CAA) who organized the Chinese Arctic Yellow River Station Expedition in 2007. We thank Jacqueline M. Grebmeier at the Center for Environmental Science of the University of Maryland for language improvement on this paper. We also are grateful to three anonymous reviewers for suggestive comments and modification. This work was supported by the National Natural Science Foundation of China (Grant no. 40676002 and 40876097) and China’s Action Plan for the International Polar Year (IPY).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zeng, Y., Zheng, T. & Li, H. Community composition of the marine bacterioplankton in Kongsfjorden (Spitsbergen) as revealed by 16S rRNA gene analysis. Polar Biol 32, 1447–1460 (2009). https://doi.org/10.1007/s00300-009-0641-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-009-0641-2