
Abstract
Key message
Insertion of a solo LTR, which possesses strong bidirectional, stem-specific promoter activities, is associated with the evolution of a dwarfing apple spur mutation.
Abstract
Spur mutations in apple scions revolutionized global apple production. Since long terminal repeat (LTR) retrotransposons are tightly related to natural mutations, inter-retrotransposon-amplified polymorphism technique and genome walking were used to find sequences in the apple genome based on these LTRs. In ‘Red Delicious’ spur mutants, a novel, 2190-bp insertion was identified as a spur-specific, solo LTR (sLTR) located at the 1038th nucleotide of another sLTR, which was 1536 bp in length. This insertion-within-an-insertion was localized within a preexisting Gypsy-50 retrotransposon at position 3,762,767 on chromosome 4. The analysis of transcriptional activity of the two sLTRs (the 2190- and 1536-bp inserts) indicated that the 2190-bp sLTR is a promoter, capable of bidirectional transcription. GUS expression in the 2190-bp-sense and 2190-bp-antisense transgenic lines was prominent in stems. In contrast, no promoter activity from either the sense or the antisense strand of the 1536-bp sLTR was detected. From ~150 kb of DNA on each side of the 2190 bp, sLTR insertion site, corresponding to 300 kb of the ‘Golden Delicious’ genome, 23 genes were predicted. Ten genes had predicted functions that could affect shoot development. This first report, of a sLTR insertion associated with the evolution of apple spur mutation, will facilitate apple breeding, cloning of spur-related genes, and discovery of mechanisms behind dwarf habit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apple trees (Malus × domestica Borkh.) are one of the most widespread fruit trees in temperate regions of the world. Since these fruit trees are naturally heterozygous, due to their self-incompatibility, and have a long-lasting juvenile phase, it is impossible to introduce genes associated with desirable traits into a commercially successful apple cultivar such as ‘Red Delicious’ through conventional breeding methods.
Mutation has proven to be one of the most important breeding methods for genetic improvement of apple cultivars. A number of apple mutants have become commercially successful cultivars, including those derived from the spontaneous spur-type or compact growth habit mutation. A spur-type growth habit results in the formation of numerous short fruit spurs, instead of large side branches, which allows higher planting density in orchards, minimizes orchard maintenance and pruning, and increases fruit yield and quality. One goal of apple breeding is to generate varieties with spur, dwarf, and semi-dwarf growth habits. Around the world, mutant spur scions are grafted onto regional apple dwarfing rootstocks to control apple tree size and shoot number.
In recent years, there have been some reports on the mechanisms behind dwarfing in apple. Three quantitative trait loci (QTLs) on chromosomes 5, 11 and 13 were reported to control apple rootstock dwarfing, namely Dw1, Dw2 and Dw3, respectively (Tustin et al. 2015; Harrison et al. 2016). While the molecular bases of dwarfing spur mutations are still unknown, the spur mutants (sports) have enabled the identification of the key player controlling specific aspects of tree architecture (Petersen and Krost 2013). The apple columnar mutation was shown to be associated with an integration of a Gypsy-like retrotransposon on chromosome 10 (Otto et al. 2014). The polymorphism between the cultivar ‘Red Delicious’ and its spur mutants was detected using sequence-specific amplification polymorphism (S-SAP) based on retrotransposon LTRs (Sun et al. 2010a).
Increasingly, retrotransposons are being identified as the molecular bases of mutations in genomes (Grandbastien 2015). For instance, the mutation leading to cold-dependent accumulation of anthocyanins in Citrus is attributed to transposition and recombination of a cold-inducible LTR retrotransposon (Butelli et al. 2012). In rice, a retrotransposon insertion resulted in the loss of function of OsMADS3, which leads to recessive male sterility (Zhang et al. 2015). An LTR retrotransposon insertion in the class IV HD-ZIP transcription factor gene CsGL3 leads to the glabrous mutation in cucumber (Pan et al. 2015).
Retrotransposons are the most abundant and widespread class of eukaryotic transposable elements and proliferate via RNA intermediates, such as retroviruses. Retrotransposons are divided into long terminal repeat (LTR) retrotransposons and non-LTR retrotransposons (Grandbastien 1998). In the apple genome, LTR retrotransposons (LRNs) are the most abundant type of transposable elements (Velasco et al. 2010). LRN activity produces changes in genes and genome structure via transposition, insertion, excision, chromosome breakage and recombination, often with accompanying alterations in gene activity (Grandbastien 2015).
The LTR segments of LRNs contain transcriptional promoter and enhancer elements, such as TATA boxes, hormone receptor-binding sites and enhancer core elements. Promoter and enhancer activities of LTRs have been shown in some plants (Kashkush and Khasdan 2007; Butelli et al. 2012; Grandbastien 2015). Thus, the LTR can be involved in the transcriptional regulation of surrounding genes. Unequal recombination or illegitimate recombination between LRNs can produce sLTRs, which lack the gag-pol genes of the retrotransposon, yet are associated with host genome evolution (Oyama et al. 2010; Kahyo et al. 2013; Zhang et al. 2015).
In previous papers, we found that apple spur mutations were closely related to polymorphism of an LTR insertion (Sun et al. 2010b). Based on this LTR in ‘Red Delicious’ spur mutants, inter-retrotransposon amplified polymorphisms (IRAP) were used to identify a specific DNA band in this study. Genome walking was used to identify a novel insertion of a 2190-bp sLTR at the 1038th nucleotide of another, 1536 bp, sLTR, which was located within a preexisting retrotransposon, Gypsy-50, at position 3,762,767 on chromosome 4 in ‘Red Delicious’ spur mutants. This is the first reported association between the insertion of a sLTR and spur mutations. Furthermore, the data show that the 2190-bp sLTR is a strong promoter, capable of bidirectional transcription, and that the bidirectional expression activities of the 2190-bp sLTR are prominent in the stem. Genes in the regions flanking the target insertion site of the sLTR on chromosome 4 are discussed as candidates for involvement in control of shoot development as it relates to the spur phenotype.
Materials and methods
Plant material
Apple varieties ‘Red Delicious’; its non-spur sports ‘Richared’, ‘Starking’, and ‘Early Red Delicious’; and its spur mutants ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, ‘Oregon spur Delicious’ and ‘Jujiadian spur starking’ were obtained from the National Malus Genus Germplasm Repository at Xingcheng, Liaoning, China and were grown at the experimental farm at Anhui Academy of Agricultural Sciences, Hefei, China. Total DNA was extracted from apple leaves using a modified method following Murray and Thompson (Murray and Thompson 1980). DNA concentration and purity were assessed by absorbance at 260 and 280 nm (Eppendorf Biophotometer, Hamburg, Germany).
IRAP technique and genome walking
Primers (Table 1), designed from RNase-LTR fragments (Sun et al. 2010a), were used to detect polymorphism fragments between Red Delicious and its spur mutants ‘Chinese Marshal 1’ and ‘Oregon spur Delicious’ using IRAP method (Sun et al. 2010b). The specific fragment obtained from the two spur mutants was named Fragment-1. The sequence flanking specific Fragment-1 was isolated by TAIL-PCR (Liu et al. 1995) from the spur mutants and named Sequence-2. The sequence flanking Sequence-2, Sequence-3, was isolated by IPCR (inverse PCR) from the two spur mutants. Primers LP-1 and LP-2 were designed based on the sequence of the apple genome to amplify the sequence flanking Sequence-3. The schematic of IRAP and genome walking is shown in Figure S1. PCR was performed with KOD FX Neo polymerase (Toyobo, Shanghai) following the manufacturer’s protocol. All primers used in this study are listed in Table 1.
Cloning, sequencing and data search
The IRAP, Tail-PCR and IPCR products were visualized on 1.5% agarose gels in 1× TAE buffer under UV light after staining with ethidium bromide. All of the fragments were recovered by Gel DNA Purification and Recovery Kit (Takara, Japan). The recovered fragments were ligated into the pGEM-T Easy vector (Takara, Japan) and transformed into competent Escherichia coli DH5α. Recombinant clones were directly used as templates for PCR, and the positive clones were selected for sequencing, which was performed by Sangon Biological Engineering and Technology Service, Shanghai, China. The 10–12 kb amplification products of primers LP-1 and LP-2 were recovered and further amplified in about 3–4 kb fragments using overlapped primers (Table S1). These fragments were cloned and sequenced. The basic local alignment search tool (BLAST) alignment was used to map the loci of the clones in the ‘Golden Delicious’ genomic sequence using Genome Database of Rosaceae (GDR) GBrowse (http://www.rosaceae.org), and the clones were further investigated with the BLAST at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). If the cloned sequence covering the junction of the specific insertion sequence in a spur mutant and both the 5′-end and 3′-end flanking genomic regions were not identified within the apple genomic database, the finding was considered to indicate a novel insertion.
Detection of the insertion polymorphism between ‘Red Delicious’ and its spur mutants
The primers LP-1 and LP-2 (Table 1) were used to detect the insertion polymorphisms among ‘Red Delicious’ and its non-spur and spur mutants. PCR was performed with KOD FX Neo as described above. The PCR products were detected by electrophoresis using 0.9% agarose.
Sequence analysis of the 2190-bp insertion
The 2190-bp insertion fragment was set as a DNA query sequence to search the apple, pear, peach, and strawberry genome databases using BLAST. Returned sequences with an identity >80% and a length >1777 bp were extracted as the target repeat.
Ectopic expression of the 2190- and 1536-bp insertion fragments
To determine if the 2190-bp insertion is associated with apple spur mutations, the bidirectional transcription of the 2190- and 1536-bp fragments were analyzed using ectopic expression. The 35S promoter in the pBI121 vector was substituted by either the 2190-bp sense strand, the 2190-bp-antisense strand, the 1536-bp sense strand, or the 1536-bp-antisense strand using In-Fusion HD Cloning Kits (Takara) to form the respective recombinant constructs (pB::sense-2190::GUS, pB::antisense-2190::GUS, pB::sense-1536::GUS and pB::antisense-1536::GUS). The fusion constructs and the positive control CaMV 35S-GUS (pBI121) were separately transferred into the Agrobacterium rhizogenes strain K599. Transgenic soybean hairy roots were generated following (Kereszt et al. 2007) by infecting cotyledonary nodes of 3–5 day-old soybean (Tianlong 8) seedlings with Agrobacterium rhizogenes K599 containing recombinant vectors. Wild-type hairy roots were generated through the procedure described above using Agrobacterium rhizogenes K599 without recombinant vectors. Antibiotic-resistant plants were identified by PCR using two pairs of primers (Table 1). GUS activity in hair roots and stems of positive plants were detected histochemically (Jefferson et al. 1987).
Candidate gene analysis at the regions flanking the 2190-bp insertion
Predicted gene transcripts within the roughly 150-kb flanking regions on either side of the 2190-bp insertion site were extracted from the ‘Golden Delicious’ genomic sequence using GDR GBrowse. Their locations and putative functions were analyzed online with UniProt (http://www.uniprot.org/).
Database searches
BLAST searches against the apple genome (Velasco et al. 2010) were carried out at the NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) or the Genome Database of Rosaceae (http://www.rosaceae.org). BLAST searches against the Arabidopsis genome were conducted using The Arabidopsis Information Resource (TAIR) (Lamesch et al. 2012). Sequences of interest were analyzed online with CENSOR (Kohany et al. 2006), which performs BLAST searches against transposon databases, and GENSCAN (Burge and Karlin 1997), which can detect open reading frames (ORFs).
Results
Insertion of a sLTR in spur mutants of ‘Red Delicious’
Genomic differences between the parent ‘Red Delicious’ and its spur mutants ‘Chinese Marshal 1’ and ‘Oregon spur Delicious’ were detected using the IRAP technique. Of the 15 pairs of primers investigated herein, one produced a specific fragment of 809 nucleotides in the two spur mutants but not in ‘Red Delicious’ (Fig. 1a; Table 1).

The polymorphism between ‘Red Delicious’ and its spur mutants. a IRAP exhibited polymorphism between ‘Red Delicious’ (R) and the spur mutants ‘Chinese Marshal 1’ (C) and ‘Oregon spur Delicious’ (O). The red arrow indicates the specific band in the spur mutants. b The specific band produced by TAIL-PCR and IPCR in spur mutants ‘Chinese Marshal 1’ (C) and ‘Oregon spur Delicious’ (O). c The polymorphism produced by primers LP-1 and LP-2 among ‘Red Delicious’ (R) and its non-spur mutants ‘Richared (Ri)’, ‘Starking (S)’, and ‘Early Red Delicious (E)’ and its spur mutants ‘Chinese Marshal 1’ (C), ‘Meiguihong’ (M), ‘Show Red’ (SR), ‘Oregon spur Delicious’ (O), and ‘Jujiadian spur starking’ (J). The green triangle indicates band-1; the blue triangle indicates the band-2; the red triangle indicates the band-3 (color figure online)
Primers designed within the 809-bp fragment were used to isolate the flanking fragments from the DNA of the two spur mutants ‘Chinese Marshal 1’ and ‘Oregon spur Delicious’ by TAIL-PCR and IPCR techniques. A 4345-bp sequence, absent in ‘Red Delicious’, was obtained from the two spur mutants (Fig. 1b). The 4345-bp sequence consisted of two portions that mapped to different regions in the apple genome: 2187 bp from the 5′-end and 2158-bp from the 3′-end (Velasco et al. 2010). The 4345-bp sequence was not identified within the apple databases (Velasco et al. 2010).
The primers LP-1 and LP-2 were designed to the regions flanking the 4345-bp insert (Table 1; Figure S1), based on the apple databases (Velasco et al. 2010), to amplify the sequences flanking the insertion site in ‘Red Delicious’, its three non-spur mutants, namely ‘Richared’, ‘Starking’, and ‘Early Red Delicious’, and its five spur mutants, namely ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, ‘Oregon spur Delicious’ and ‘Jujiadian spur starking’ (Lu and Jia 1999) (Fig. 1c). ‘Red Delicious’ carried band-2 and band-3, which were the two shortest bands (Figs. 1c, 3a). The results suggested that the genomic sequences amplified using LP-1 and LP-2 are different between the non-spur cultivars and the spur mutants. In Fig. 1c, band-1 was obtained from five spur mutants: ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, ‘Oregon spur Delicious’ and ‘Jujiadian spur starking’. Band-2 was obtained from both the non-spur cultivars and the spur mutants, except for ‘Oregon spur Delicious’. Band-3 was obtained from the non-spur cultivars and the spur mutant ‘Oregon spur Delicious’. Band-1, band-2 and band-3 were recovered, sequenced, and mapped to the apple genome (Velasco et al. 2010).
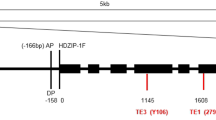
Band-2, 8329 bp in length, was mapped to chromosome 4 between positions 3,762,441 and 3,770,754. The sequence structure of band-3 (6758 bp in length) revealed that a 1536-bp fragment was deleted from band-2 at position 3,767,540 (Figs. 2, 3). The sequence structure of band-1 (10,504 bp in length) revealed that a 2190-bp fragment was inserted within the 1536-bp sequence at nucleotide 1038 in band-2 (2190 bp was at position 5100 in the 8329-bp fragment and at position 3,768,577 on chromosome 4) (Figs. 2, 3, Figure S2). The 2190-bp sequence was inserted into the 1536-bp band in the five spur mutants, but not present in the non-spur cultivars at this position of chromosome 4 (Figs. 1c, 2, 3). This analysis showed that it is not a 2190-bp deletion in ‘Red Delicious’ that gave rise to the spur mutants, but rather an insertion. The spur mutations are associated with the insertion of the 2190-bp fragment.

The 8329-bp region on chromosome 4 in non-spur type mutants (NS) and spur-type mutants (S). The grey represents the protein region and the purple boxes represents the long terminal repeat (LTR) of the retrotransposon Gypsy-50 (from nucleotide 3,762,767 to 3,770,535 on chromosome 4). The green box represents the 1536-bp sLTR (from 3,767,540 bp to 3,769,063 bp). The red box represents the 2190-bp insertion at the 1038th nucleotide (red triangle) of the 1536-bp sLTR in spur-type mutants. The target site duplications (TSD) and inverted repeats (IR) of Gypsy-50, 1536-bp, and 2190-bp sLTRs are indicated in purple, green, and red, respectively (color figure online)

Structural maps of the Gypsy-50 retrotransposon on chromosome 4 in the different apple varieties. The grey and purple boxes represent the protein region and the long terminal repeats (LTRs), respectively, of the retrotransposon Gypsy-50 that starts at position 3,762,767. The green box represents the 1536-bp sLTR at position 4774 of the Gypsy-50 LTN. The red box represents the 2190-bp insertion at the 1038th nucleotide site of the 1536-bp sLTR. a The two alleles in cultivars ‘Red Delicious’, ‘Richared’, ‘Starking’, and ‘Early Red Delicious’. b The two alleles present in cultivars ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, and ‘Jujiadian spur starking’. c The two alleles in the culticar ‘Oregon spur Delicious’ (color figure online)
Band-2, containing the 1536-bp fragment, and band-3, missing the 1536-bp fragment, were both amplified from ‘Red Delicious’ and its non-spur mutants ‘Richared’, ‘Starking’, and ‘Early Red Delicious’ (Figs. 1c, 3a). Sequencing of multiple amplicons revealed that the 1536-bp fragmentat position 3,767,540 on chromosome 4 was heterozygous in ‘Red Delicious’ and its non-spur mutants, i.e., one allele contained the extra 1536 bp, which was deleted in the other allele (Fig. 3a).
In the spur mutants, there were two amplified bands. Band-1, containing both the 2190- and the 1536-bp insertions, and band-2, containing only the 1536-bp insertion, were both amplified in the accessions ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, and ‘Jujiadian spur starking’ (Figs. 1c, 3b). Sequence analysis of band-1 and band-2 showed that the 2190- and 1536-bp fragments could be co-integrated at position 3,767,540 on chromosome 4 (Fig. 3a, b). Furthermore, the analysis revealed that the 2190-bp fragment was inserted into the 1536-bp fragment at nucleotide 1038.
A third pattern was only seen in the spur mutant ‘Oregon spur Delicious’. Band-1, containing both the 2190- and 1536-bp insertions, and band-3, without any insertion, were both amplified from accession ‘Oregon spur Delicious’. The presence of the 10,504-bp fragment was heterozygous, i.e., one allele contained the 2190-bp insert in the 1536-bp fragment, but the other allele lacked both the 1536- and the 2190-bp insertions (Figs. 1c, 3c).
Together, the above results showed that the 2190-bp insert in the 1536-bp sequence at position 3,767,540 on chromosome 4 induced the apple spur mutation and that the 2190-bp insertion was associated with the 1536-bp insert in the spur mutants.
Sequence analysis of the 2190- and 1536-bp insertions
The sequences of bands-1, -2, and -3 were analyzed in detail. Interestingly, when the 2190-bp sequence was inserted into the 1536-bp insert at the 1038th nucleotide site in the spur mutants, the 5-bp target site duplication (CGGGG) next to the left insertion site was copied to the right insertion site (Fig. 2, Figure S2). That is, duplicated CGGGG motifs flanked the 2190-bp insertion. The 2190-bp fragment contained the 5′-terminal and 3′-terminal sequences 5′-TGAT…ATCA-3′ (Fig. 2, Figure S2). The 1536 bp was flanked by 5-bp direct complete repeats (ATTCA at the 5′- and 3′-ends), representing target-site duplication, and contained the 5′-terminal and 3′-terminal sequences 5′-TGTA…TACA-3′ (Fig. 2, Figure S2). These are typical of the LTRs of LRNs and insertion sites (Vitte and Panaud 2003). The 2190- and 1536-bp sequences contained no predicted open reading frames to encode the proteins required for transposition. The 2190- and 1536-bp fragments were identified as sLTRs.
The novel insertion that induced spur mutations was a sLTR 2190-bp in length. CENSOR analysis (Kohany et al. 2006) of the immediate flanking regions of the target site of the sLTR insertion revealed that the 2190-bp LTR was inserted at the 1038th nucleotide site of 1536-bp, which itself was localized within a preexisting Gypsy-like retrotransposon, Gypsy-50, at position 3,762,767 on apple chromosome 4 (Fig. 2).
We searched for the 2190-bp sequence in the apple, pear, peach, and strawberry genomic sequence databases. There are 122 copies of the 2190-bp insertion sequence in apple (Malus × domestica, identity >80%) with lengths ranging from 1777- to 2190-bp. DNAMAN was used for Multiple Alignment analysis of the 2190-bp insertion sequences. The overall identity of the copies from apple was 74.91% (Figure S2). No homologous copies were found in strawberry or peach. In pear, the sizes of the similar copies were about 1000 bp and shared a conserved middle region.
Predicted genes in the region interrupted by the 2190-bp insertion on chromosome 4
To understand the impact of the 2190-bp insertion on the expression levels of genes flanking the insertion site on chromosome 4, ORFs were sought in about 150 kb of the genomic sequence flanking the insertion site. A total of 23 putative ORFs were found. Among the gene functions coded by the putative ORFs, one acyl-coenzyme A oxidase 1 (ACX1), one ethylene-responsive transcription factor (ERF), several auxin-induced proteins 15A, an auxin-responsive protein SAUR, several MYB transcription factors, a 1-aminocyclopropane-1-carboxylate oxidase (ACO), several cyclin-dependent kinases, a CBL-interacting protein kinase, a 26S proteasome non-ATPase regulatory subunit, and a E3 ubiquitin-protein ligase were identified.
The apple 2190 bp, sLTR is a strong promoter and functions efficiently in two directions in stems
To further characterize the promoter functions of the 2190- and 1536-bp insertions, we made several constructions in which the sense or antisense strand of the 2190-bp fragment and the sense or antisense strand of the 1536-bp fragment were fused to the GUS reporter gene in the plasmid pBI-121. The constructs were transformed into soybean hairy roots using the A. rhizogenes K599 strain. Antibiotic-resistant plants were confirmed by PCR using two pairs of primers (Table 1); only those yielding the expected PCR fragments by both primers were regarded as positive. GUS activity in hairy roots and stems of positive plants were detected histochemically (Fig. 4). Hairy roots and stems from lines carrying the 1536-bp-sense or 1536-bp-antisense constructs did not show GUS staining (Fig. 4c, d); whereas hairy roots and stems carrying either the 2190-bp-sense and 2190-bp-antisense constructs did show GUS staining in histochemical assays. GUS expression in the 2190-bp-sense and 2190-bp-antisense lines were prominent in stems (Fig. 4e, f). Thus, the 2190-bp, sLTR is a stronger bidirectional promoter, while the 1536-bp, sLTR has no detectable promoter function.

Histochemical analysis of the promoter activities of the sense-strand (sense-2190:GUS) and the antisense-strand (antisense-2190:GUS) of the 2190-bp sLTR and the sense-strand (sense-1536:GUS) and the antisense-strand (antisense-1536:GUS) of the 1536-bp sLTR in transgenic soybean hairy roots. a Wild-type hairy roots. b CaMV 35S:GUS-transformed hairy roots. c sense-2190:GUS-transformed hairy roots. d antisense-2190:GUS-transformed hairy roots. e sense-1536:GUS-transformed hairy roots. f antisense-1536:GUS-transformed hairy roots
Discussion
Inter-retrotransposon amplified polymorphism (IRAP) and genome walking were used to identify a novel insertion of a sLTR, 2190 bp in length that is associated with apple spur mutants. Future combination of molecular marker technology based on retrotransposon LTR and genome walking could lead to identification of more polymorphic loci in somatic mutants that contain sLTRs or retrotransposons. Because the activity of retrotransposons can cause genomic variations, understanding associations between LTNs and spur mutants may lead to new insights into bud mutations (Butelli et al. 2012; Kahyo et al. 2013; Zhang et al. 2015).
The molecular analysis of the apple spur mutants indicated that the spur mutations are related to the insertion of the 2190-bp sLTR at the 1038th site in another sLTR, a 1536 bp at position 3,767,540 on chromosome 4 (Fig. 2). It is unclear precisely how the spur phenotype could be generated by the 2190-bp insertion. Retrotransposons tend to insert into each other at transposon-rich regions, similar to our observation that the spur-specific, sLTR (2190 bp) is located within another sLTR (1536 bp), which is localized within a preexisting Gypsy-50 retrotransposon (Fig. 2, Figure S2).
Confusingly, the 2190-bp insertion occurred only in the 1536 bp, which contains only one targeting insertion site (the ‘CGGGG’ of the 2190-bp LTR), although there were two ‘CGGGG’ sites in Gypsy-50 (Figure S2). The insertion of the 2190-bp sLTR could have been accompanied by the insertion of the 1536-bp sLTR on a chromosome 4 that did not contain either sLTR, for instance, in accessions ‘Chinese Marshal 1’, ‘Meiguihong’, ‘Show Red’, and ‘Jujiadian spur starking’. On the other hand, in ‘Oregon spur Delicious’, the 2190-bp sLTR was directly inserted into the 1536-bp sLTR (Fig. 3).
It is well known that LTRs of retrotransposons are functional as promoters (Butelli et al. 2012; Grandbastien 2015), similar to our results that the 2190-bp sLTR is a stronger promoter, capable of bidirectional transcription when introduced into soybean by transfection (Fig. 4). It is surprising that the bidirectional expression activities of the 2190-bp sense and antisense strands were prominent in stems, which suggested that the genes controlling spur phenotype were regulated through temporal spatial expression of the 2190-bp promoter. However, the mechanism underlying this stem-prominent expression of the 2190-bp bidirectional promoter remains unknown, highlighting the need for future detailed analysis of the 2190-bp bidirectional promoter. In contrast, promoter function of the 1536-bp sLTR could not be detected, suggesting that the insertion of the 1536-bp sLTR does not influence the expression of flanking sequences. However, the stronger bidirectional transcription driven by the 2190-bp insert could significantly influence expression of flanking genes (Hayashi and Yoshida 2009; Butelli et al. 2012).
It has been shown that inserted transposons can influence expression of genes that are localized dozens of kilobases downstream of the insertion sites. For instance, Gypsy-44, inserted in the Co region, can significantly upregulate the level of transcription of the gene MDP0000934866, which is located approximately 90 kb downstream of the Gypsy-44 insertion in the apple columnar mutant (Otto et al. 2014). Similarly, the insertion of a Hopscotch retrotransposon 60 kb upstream of the teosinte branched 1 (tb1) locus enhances expression of tb1 in maize, which leads to a severe reduction of branching in maize compared to its wild progenitor teosinte (Studer et al. 2011). Thus, the regions flanking the insertion site, roughly 150-kb on either side, were used to predict genes based on the apple genome. A total of 23 putative genes were identified. Several potentially interesting candidate genes were found in the physical genomic region fully associated with the spur mutation (Table 1). Genes of particular interest included acyl-coenzyme A oxidase 1 (ACX1), ethylene-responsive transcription factor ERF, 1-aminocyclopropane-1-carboxylate oxidase (ACO), auxin-induced protein15A, auxin-responsive protein SAUR, MYB, pentatricopeptide repeat-containing protein, cyclin-dependent kinase, and CBL-interacting protein kinase. These candidate genes, selected by potential function related to the spur trait, could be influenced by the insertion of the strong, bidirectional-activating, 2190-bp sLTR. The biosynthesis of jasmonic acid (JA) in plant peroxisomes requires the action of acyl-coenzyme A oxidase 1 (ACX 1) (Koo et al. 2006; Schilmiller et al. 2007), and JA is involved in the regulation of plant dwarfing (Światek et al. 2004). The ethylene-responsive transcription factor ERF belongs to one of the most important gene families in plants, which plays the vital role of regulating plant growth and development as well as response to diverse stresses (Cui et al. 2016). In rice, the ERF OsEATB was identified to restrict internode elongation by down-regulating a gibberellin biosynthetic gene (Qi et al. 2011). Ethylene regulates essentially all physiological processes during the plant’s life cycle and is responsible for signaling changes in shoot and leaf formation, flower and fruit development, organ senescence and abscission, plant defense mechanisms, and a number of interactions with other plant hormones. ACC oxidase (ACO) is involved in the final step of ethylene production in plant tissues (Ruduś et al. 2013). The phytohormone auxin regulates numerous aspects of plant growth and development and can exert rapid and specific effects on genes at the molecular levels. Overexpression of the KNOX gene Tkn4 in tomato results in a dwarf phenotype in which the expression level of auxin-induced protein 15A was upregulated (Yan et al. 2015). The early auxin-responsive genes SAURs are key effector outputs of hormonal and environmental signals that regulate plant growth and development (Ren and Gray 2015). MYB transcription factors are involved in many physiological processes, such as shoot morphogenesis and leaf patterning (Guo et al. 2008), secondary metabolism (Yang et al. 2012) and anthocyanin accumulation (Chagné et al. 2013). Pentatricopeptide repeat (PPR) proteins are RNA-binding proteins that have a range of essential functions in post-transcriptional processes (including RNA editing, RNA splicing, RNA cleavage and translation) within mitochondria and chloroplasts and have profound effects on organelle biogenesis and function and, consequently, on photosynthesis, respiration, plant development, and environmental responses (Barkan and Small 2014). Cyclin-dependent kinase plays important roles involved in the integration of extracellular and intracellular signals to modulate gene transcription and cell division (Malumbres et al. 2014). In Arabidopsis, CBL-interacting protein kinase (CIPK) is involved in response to the plant hormone ABA and different stresses (Hashimoto and Kudla 2011). Genes belonging to any one of the above-mentioned families could be considered putative candidate genes controlling spur habit in apple. Moreover, the strong bidirectional promoter within the 2190-bp insert could activate multiple flanking candidate genes that together give rise to a common spur phenotype.
In conclusion, we used IRAP and genome walking techniques to identify an insertion mutation of a sLTR, 2190-bp in length, in apple spur mutants. This discovery provided not only new insights into the genetic mechanism behind spur mutants but also new ways to identify bud mutations. The 2190-bp sLTR is a strong bidirectional stem-specific promoter localized at the 1038th nucleotide of another 1536 bp, sLTR, which lacked apparent transcriptional activities. This sLTR was inserted at position 3,768,577 on chromosome 4. According to the genomic sequence of ‘Golden Delicious’, about 150 kb on either side of the 2190-bp insertion putatively contain 23 genes, including at least ten genes coding for protein functions that could be responsible for the spur-habit trait. These results are valuable for future identification of the mechanisms behind apple spur mutation. To identify reliable candidate genes and perhaps find the mis-expressed gene responsible for the spur habit, a more extensive analysis that includes the functional identification of the candidate genes is still required.
Author contribution statement
JS and QS conceived the study, designed the experiments and interpretation of results; MH, JZ, HQ, JG, LL and WM performed experiments; JS, QS, MH and JZ analyzed data and wrote this manuscript. All authors read and approved the manuscript.
Abbreviations
- ACX1:
-
Acyl-coenzyme A oxidase 1
- ACO:
-
1-Aminocyclopropane-1-carboxylate oxidase
- CIPK:
-
CBL-interacting protein kinase
- ERF:
-
Ethylene-responsive transcription factor
- IRAP:
-
Inter-retrotransposon amplified polymorphism
- LTR:
-
Long terminal repeat
- LRNs:
-
LTR retrotransposons
- ORFs:
-
Open reading frames
- QTL:
-
Quantitative trait loci
- IPCR:
-
Reverse PCR
- S-SAP:
-
Sequence-specific amplification polymorphism
- sLTR:
-
Solo LTR
- TAIL-PCR:
-
Thermal asymmetric interlaced PCR
References
Barkan A, Small I (2014) Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol 65:415–442. doi:10.1146/annurev-arplant-050213-040159
Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268:78–94
Butelli E, Licciardello C, Zhang Y et al (2012) Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24:1242–1255. doi:10.1105/tpc.111.095232
Chagné D, Lin-Wang K, Espley RV et al (2013) An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol 161:225–239. doi:10.1104/pp.112.206771
Cui L, Feng K, Wang M et al (2016) Genome-wide identification, phylogeny and expression analysis of AP2/ERF transcription factors family in Brachypodium distachyon. BMC Genomics 17:636. doi:10.1186/s12864-016-2968-8
Grandbastien MA (1998) Activation of plant retrotransposons under stress conditions. Trends Plant Sci 3:181–187. doi:10.1016/S1360-1385(98)01232-1
Grandbastien MA (2015) LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim Biophys Acta Gene Regul Mech 1849:403–416. doi:10.1016/j.bbagrm.2014.07.017
Guo M, Thomas J, Collins G, Timmermans MCP (2008) Direct repression of KNOX loci by the ASYMMETRIC LEAVES1 complex of Arabidopsis. Plant Cell 20:48–58. doi:10.1105/tpc.107.056127
Harrison N, Harrison RJ, Barber-perez N et al (2016) A new three-locus model for rootstock-induced dwarfing in apple revealed by genetic mapping of root bark percentage. J Exp Bot 67:1871–1881. doi:10.1093/jxb/erw001
Hashimoto K, Kudla J (2011) Calcium decoding mechanisms in plants. Biochimie 93:2054–2059. doi:10.1016/j.biochi.2011.05.019
Hayashi K, Yoshida H (2009) Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J 57:413–425. doi:10.1111/j.1365-313X.2008.03694.x
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907. doi:10.1073/pnas.1411926112
Kahyo T, Tao H, Shinmura K et al (2013) Identification and association study with lung cancer for novel insertion polymorphisms of human endogenous retrovirus. Carcinogenesis 34:2531–2538. doi:10.1093/carcin/bgt253
Kashkush K, Khasdan V (2007) Large-scale survey of cytosine methylation of retrotransposons and the impact of readout transcription from long terminal repeats on expression of adjacent rice genes. Genetics 177:1975–1985. doi:10.1534/genetics.107.080234
Kereszt A, Li D, Indrasumunar A et al (2007) Agrobacterium rhizogenes-mediated transformation of soybean to study root biology. Nat Protoc 2:948–952. doi:10.1038/nprot.2007.141
Kohany O, Gentles AJ, Hankus L, Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform 7:474. doi:10.1186/1471-2105-7-474
Koo AJK, Hoo SC, Kobayashi Y, Howe GA (2006) Identification of a peroxisomal acyl-activating enzyme involved in the biosynthesis of jasmonic acid in Arabidopsis. J Biol Chem 281:33511–33520. doi:10.1074/jbc.M607854200
Lamesch P, Berardini TZ, Li D et al (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40:1202–1210. doi:10.1093/nar/gkr1090
Liu YG, Mitsukawa N, Whittier RF (1995) Efficient isolation and map-ping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Lu QN, Jia DX (1999) Malus Mill Flora, China fruit-plant monographs. China Agricultural Science and Technology Publishing House, China Forestry Publishing House, Beijing
Malumbres M, Manning G, Whyte D et al (2014) Cyclin-dependent kinases. Genome Biol 15:122. doi:10.1186/gb4184
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4325
Otto D, Petersen R, Brauksiepe B et al (2014) The columnar mutation (“Co gene”) of apple (Malus × domestica) is associated with an integration of a Gypsy-like retrotransposon. Mol Breed 33:863–880. doi:10.1007/s11032-013-0001-3
Oyama RK, Silber MV, Renner SS (2010) A specific insertion of a solo-LTR characterizes the Y-chromosome of Bryonia dioica (Cucurbitaceae). BMC Res Notes 3:166. doi:10.1186/1756-0500-3-166
Pan Y, Bo K, Cheng Z, Weng Y (2015) The loss-of-function GLABROUS 3 mutation in cucumber is due to LTR-retrotransposon insertion in a class IV HD-ZIP transcription factor gene CsGL3 that is epistatic over. BMC Plant Biol 15:302. doi:10.1186/s12870-015-0693-0
Petersen R, Krost C (2013) Tracing a key player in the regulation of plant architecture: the columnar growth habit of apple trees (Malus × domestica). Planta 238:1–22
Qi W, Sun F, Wang Q et al (2011) Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol 157:216–228. doi:10.1104/pp.111.179945
Ren H, Gray WM (2015) SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol Plant 8:1153–1164. doi:10.1016/j.molp.2015.05.003
Ruduś I, Sasiak M, Kepczyński J (2013) Regulation of ethylene biosynthesis at the level of 1-aminocyclopropane-1-carboxylate oxidase (ACO) gene. Acta Physiol Plant 35:295–307. doi:10.1007/s11738-012-1096-6
Schilmiller AL, Koo AJK, Howe GA (2007) Functional diversification of acyl-coenzyme A oxidases in jasmonic acid biosynthesis and action. Plant Physiol 143:812–824. doi:10.1104/pp.106.092916
Studer A, Zhao Q, Ross-Ibarra J, Doebley J (2011) Identification of a functional transposon insertion in the maize domestication gene tb1. Nat Genet 43:1160–1163. doi:10.1038/ng.942
Sun J, Fang JG, Wang F et al (2010a) Characterisation of RNaseH-LTR sections of Ty1-copia retrotransposons in apple and fingerprinting of four apple clones by S-SAP analysis. J Hortic Sci Biotechnol 85:53–58. doi:10.1080/14620316.2010.11512630
Sun J, Zhou J, Sun Q, Wang K (2010b) IRAP identification of apple red delicious and Fuji bud sport clones. Acta Bot Boreal Occident 10:1952–1958
Światek A, Azmi A, Stals H et al (2004) Jasmonic acid prevents the accumulation of cyclin B1;1 and CDK-B in synchronized tobacco BY-2 cells. FEBS Lett 572:118–122. doi:10.1016/j.febslet.2004.07.018
Tustin DS, Gardiner SE, Foster TM et al (2015) Two quantitative trait loci, Dw1 and Dw2, are primarily responsible for rootstock-induced dwarfing in apple. Hortic Res 2:15001. doi:10.1038/hortres.2015.1
Velasco R, Zharkikh A, Affourtit J et al (2010) The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 42:833–839. doi:10.1038/ng.654
Vitte C, Panaud O (2003) Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol 20:528–540. doi:10.1093/molbev/msg055
Yan F, Hu G, Ren Z et al (2015) Ectopic expression a tomato KNOX Gene Tkn4 affects the formation and the differentiation of meristems and vasculature. Plant Mol Biol 89:589–605. doi:10.1007/s11103-015-0387-x
Yang C-Q, Fang X, Wu X-M et al (2012) Transcriptional regulation of plant secondary metabolism. J Integr Plant Biol 54:703–712. doi:10.1111/j.1744-7909.2012.01161.x
Zhang L, Mao D, Xing F et al (2015) Loss of function of OsMADS3 via the insertion of a novel retrotransposon leads to recessive male sterility in rice (Oryza sativa). Plant Sci 238:188–197. doi:10.1016/j.plantsci.2015.06.007
Acknowledgements
This work was supported by two Grants-in-Aid for Scientific Research from the State National Natural Science Foundation of China (Nos. 31372043 and 31071776).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Dr. Prakash P. Kumar.
Electronic supplementary material
Below is the link to the electronic supplementary material.
299_2017_2160_MOESM1_ESM.pdf
Supplementary material 1 (PDF 105 kb) The schematic of IRAP and genome walking in ‘Red Delicious’ and its two spur mutants, ‘Chinese Marshal 1’ and ‘Oregon spur Delicious’. Red box represents Fragment-1 from the two spur mutants amplified using IRAP technique. Green box represents Sequence-2 from the two spur mutants amplified using TAIL-PCR technique. Yellow box represents Sequence-3 amplified from the two spur mutants using IPCR. The primers LP-1 and LP-2 were designed based on the sequence of the apple genome to amplify the sequence flanking Sequence-1, 2 and 3
299_2017_2160_MOESM2_ESM.pdf
Supplementary material 2 (PDF 65 kb) The target site duplication (TSD, red) and inverted repeat (IR, italics) of the 1536-bp sLTR insertion (green) are shown. The TSD and IR of the 2190-bp sLTR insertion (yellow) are boxed and underlined, respectively. The TSD and IR of Gypsy-50 are indicated in red and double underlined. LTRs of Gypsy-50 are indicated in blue. The two ‘CGGGG’ sites are shaded in blue
299_2017_2160_MOESM3_ESM.png
Supplementary material 3 (PNG 4740 kb) The alignment of the 122 LTR-like copies found in apple using the 2190-bp fragment, ranging in size from 1777-bp to 2190-bp
299_2017_2160_MOESM4_ESM.pdf
Supplementary material 4 (PDF 86 kb) Primers used for overlapped amplification of products of PL-1 and PL-2, amplified by primers LP-1 and LP-2
Rights and permissions
About this article

{kind=link}
Cite this article
Han, M., Sun, Q., Zhou, J. et al. Insertion of a solo LTR retrotransposon associates with spur mutations in ‘Red Delicious’ apple (Malus × domestica). Plant Cell Rep 36, 1375–1385 (2017). https://doi.org/10.1007/s00299-017-2160-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-017-2160-x