
Abstract
Key message
A method based on DNA single-strand conformation polymorphism is demonstrated for effective genotyping of CRISPR/Cas9-induced mutants in rice.
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) has been widely adopted for genome editing in many organisms. A large proportion of mutations generated by CRISPR/Cas9 are very small insertions and deletions (indels), presumably because Cas9 generates blunt-ended double-strand breaks which are subsequently repaired without extensive end-processing. CRISPR/Cas9 is highly effective for targeted mutagenesis in the important crop, rice. For example, homozygous mutant seedlings are commonly recovered from CRISPR/Cas9-treated calli. However, many current mutation detection methods are not very suitable for screening homozygous mutants that typically carry small indels. In this study, we tested a mutation detection method based on single-strand conformational polymorphism (SSCP). We found it can effectively detect small indels in pilot experiments. By applying the SSCP method for CRISRP-Cas9-mediated targeted mutagenesis in rice, we successfully identified multiple mutants of OsROC5 and OsDEP1. In conclusion, the SSCP analysis will be a useful genotyping method for rapid identification of CRISPR/Cas9-induced mutants, including the most desirable homozygous mutants. The method also has high potential for similar applications in other plant species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a sequence-specific nuclease (SSN), the clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated 9 (Cas9) system (Cong et al. 2013; Jinek et al. 2012; Mali et al. 2013) has been recently used for genome editing in many organisms and cell lines. In this system, Cas9 protein is loaded with a synthetic single-guide RNA (sgRNA), which is a fusion of a CRISPR RNA (crRNA) and a trans-acting crRNA (tracrRNA) (Jinek et al. 2012). CRISPR/Cas9 induces a DNA double-strand break (DSB) by targeting an approximately 20 base-pair (bp) sequence upstream of a protospacer adjacent motif (PAM). The resulting DSB site is 3 bp upstream of the PAM (Jinek et al. 2012). Subsequent DNA repair by the error-prone non-homologous end-joining (NHEJ) pathway may result in mutations at the target site. Although multiple orthogonal CRISPR/Cas9 systems have been developed (Esvelt et al. 2013; Hou et al. 2013; Ran et al. 2015), Streptococcus pyogenes Cas9 (SpCas9) is prevalently used in genome editing applications presumably due its well-characterized PAM motif (NGG) (Doudna and Charpentier 2014; Hsu et al. 2014; Sander and Joung 2014).
SSNs such as zinc finger nuclease (ZFN) (Carroll 2011; Urnov et al. 2010), transcriptional activator-like effector nuclease (TALEN) (Christian et al. 2010; Li et al. 2011; Miller et al. 2011) and meganuclease (Paques and Duchateau 2007; Smith et al. 2006) all require engineering of DNA binding domains, which is not a trivial process (Gaj et al. 2013). In comparison, target recognition is simply based on DNA–RNA hybridization in the CRISPR/Cas9 system (Jinek et al. 2012), which makes it the first choice of SSNs for genome editing. However, it was found that 64–75 % of CRISPR/Cas9-generated mutations are 1 bp Indels in the model plants rice (Ma et al. 2015; Zhang et al. 2014) and Arabidopsis (Feng et al. 2014). Since CRISPR/Cas9 generates blunt-ended DNA DSBs (Jinek et al. 2012; Sternberg et al. 2014), it is not surprising this system generates a higher proportion of small indels than ZFN and TALEN which create sticky ends of 4 bp single-strand overhangs prone to degradation (Cermak et al. 2011; Kim et al. 1996). Thus, sensitive detection of very small indels is required for successful application of CRISPR/Cas9 in genome editing.
Single-strand conformational polymorphism (SSCP) analysis was first described as an efficient method for the detection of DNA polymorphisms between two alleles at chromosomal loci (Orita et al. 1989a, b). It is based on the principle that conformational difference of single-stranded DNA due to difference at the sequence level (e.g., single nucleotide polymorphisms or SNPs) can be resolved by electrophoresis in a non-denaturing gel. Coupled with polymerase chain reaction (PCR), SSCP has been used for applications such as genotyping SNPs in a crop plant (Shirasawa et al. 2004) and analysis of genetic diseases in humans (Kakavas et al. 2008).
Recently, a polyacrylamide gel electrophoresis (PAGE)-based method was used for detecting mutations generated by TALEN (Ota et al. 2013) and CRISPR/Cas9 (Zhu et al. 2014). This method is based on slower migration of heteroduplex DNA (with mismatch) than homoduplex DNA (without mismatch) in a PAGE gel. This method has limited ability to measure mutation frequency and may be unable to identify individuals which contain homozygous mutations. CRISPR/Cas9 has been shown to be highly effective for inducing targeted mutations in rice where homozygous mutants can be readily obtained in one generation (Endo et al. 2015; Feng et al. 2013; Ikeda et al. 2015; Jiang et al. 2013; Lowder et al. 2015; Ma et al. 2015; Miao et al. 2013; Mikami et al. 2015; Shan et al. 2013; Xie et al. 2015; Xu et al. 2015; Zhang et al. 2014; Zhou et al. 2014). Many of the current mutant screen methods lack the ability to identify homozygous mutants prior to Sanger DNA sequencing. Since SSCP is a powerful tool for detection of small indels (Sekiya 1996) and it is based on single-strand DNA conformation, we reasoned SSCP can be a useful genotyping tool with SSNs such as CRISPR/Cas9 because it can distinguish the wild type, heterozygous mutants and homozygous mutants. In this study, we optimized parameters for the SSCP analysis and then applied this method for screening CRISPR/Cas9-induced mutants in T0 plants of rice, which is a model plant and important crop. Mutations as small as 1-bp indels along with large indels were successfully detected by SSCP, demonstrating the SSCP-based method may broadly facilitate mutant screen in plant genome editing and beyond.
Materials and methods
Plasmid construction
The pBlueScript-derived constructs used in this study are kind gifts from Satoshi Ota and Atsuo Kawahara (Laboratory for Cardiovascular Molecular Dynamics, Quantitative Biology Center, RIKEN). To generate the Cas9 targeting construct for rice endogenous genes, the annealed gRNA oligonucleotide pair each with designed recognition sequence, was cloned into the region between the OsU6 promoter and the gRNA scaffolds of Cas9 expression backbone vector (pZHY988).
Agrobacterium-mediated rice transformation
Agrobacterium-mediated rice transformation was based on a method described previously (Hiei et al. 1994). First, dehusked seeds were briefly sterilized and cultured on solid medium at 28 °C under the dark in the growth chamber for 2–3 weeks. Actively growing calli were immersed in an Agrobacterium suspension harboring the CRISPR/Cas9 expression plasmid and transferred onto screening medium containing 50 mg/l hygromycin and 500 mg/l carbenicillin. Calli were then placed in a growth chamber for 5 weeks. During the screening stage, infected calli were transferred onto fresh screening medium every 2 weeks. After the screening stage, actively growing calli were moved onto regenerative medium for 3–4 weeks. Transgenic seedlings were then transferred into sterile plastic containers containing fresh solid medium and grown for 2–3 weeks before being transferred into soil.
Genomic DNA preparation
To detect Cas9 activity at an endogenous locus, genomic DNA was isolated from transformed rice protoplasts 2 days after transformation by following the Hexadecyltrimethylammonium bromide (CTAB) DNA extraction method (Stewart and Via 1993). Leaves of these plants were collected for DNA extraction with the CTAB method to genotype stably transformed T0 rice. Either the rice protoplasts or grinded leaves were incubated in the CTAB extraction buffer for 30 min at 65 °C. After extracting twice with a chloroform/isoamyl alcohol (24:1) mix, supernatant was collected and a tenth volume of 3 M NaAc was added. The precipitated DNA was washed with 100 % ethanol for 2 h at −20 °C. Precipitated DNA pellets were resuspended with ddH2O for the PCR-SSCP analysis.
PCR-SSCP analysis
PCR was performed using the primer pairs listed in Table S1. A typical 25 ul PCR mix contains 1 U Taq DNA Polymerase, 0.5 mM forward and reverse primer pairs, 1.5 mM MgCl2, 200 mM dNTP mix and genomic DNA template (~50 ng) or a TA cloning plasmid (~1 ng). The standard PCR condition was as follows: 94 °C for 5 min; 94 °C for 30 s, 56 °C for 30 s, 72 °C 30 s for 32 cycles; 72 °C for 5 min; hold at 10 °C. PCR products were denatured for 5 min at 95 °C and immediately put in an ice box afterwards to minimize self-annealing. Denatured PCR amplicons were electrophoresed on 15 % non-denaturing polyacrylamide gels (acrylamide–bisacrylamide; 29:1, w/w). After 6 h of electrophoresis at 45 mA, 120–200 V, polyacrylamide gels were dyed using argentation. First, the gels were stained using 0.1 % AgNO3 (100 ml volume with 0.1 g AgNO3 and 200 µl 37 % CH2O) for 10 min, developed using 2.5 % NaOH (100 ml volume with 2.5 g NaOH, 400 μl 37 %CH2O and 1 ml 4 % Na2CO3) for 10 min and then the developing process was stopped with water.
TA cloning and sequencing analysis
Targeted genomic regions were amplified by PCR. All PCR primer sequences are listed in Supplementary Table 1. All PCR products were resolved on 1 % agarose gels. Selected PCR products were excised, purified, and cloned into the pMDC18 T-vector. The targeted NHEJ events were identified by Sanger sequencing of individual clones.
Results
Position of small indels impacts resolution of the SSCP analysis
CRISPR/Cas9 cleaves the double-stranded DNA about 3 bp upstream of the PAM motif (Jinek et al. 2012) and small indels are expected to be generated at this region. When mutated DNA is amplified by PCR and denatured, we expect to detect the mutations with the SSCP analysis because mutated single-strand DNA will migrate differently compared to the wild type (WT) in non-denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. 1). We first wanted to examine how the length and relative positions of the mutations of the PCR amplicons could impact the detection in the SSCP analysis. We generated a series of pBlueScript plasmids which contain deletions of 1, 2, 5 and 9 bp compared to the WT control. We then performed PCR with eight different primer sets, which resulted in 40 sets of amplicons of different lengths with the small deletions at different relative positions (Fig. 2a). These PCR amplicons were denatured and run in a 15 % non-denaturing PAGE to evaluate the migration of single-strand DNA among all samples. We found the best resolution could be achieved when the PCR amplicons are smaller than 300 bp and the mutation sites are close to the middle of the amplicons (Fig. 2b). Under such conditions, all sizes of deletions could be distinguished from the WT. Double-strand DNA was also detected in every sample due to partial re-annealing. Because the double-stranded DNA runs much faster than the single-stranded DNA, both types of DNA can be easily distinguished (Fig. 2b). Although we only tested deletions in this experiment, insertions in principle should also be detected by the SSCP analysis.

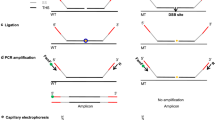
Schematic diagram of an SSCP-based genotyping method for CRISPR/Cas9-mediated mutagenesis. In this procedure, CRISPR/Cas9-induced NHEJ mutations, along with the WT DNA, are amplified by PCR. The PCR amplicons are subsequently denatured and single-strand conformation polymorphisms (SSCPs) can be resolved by running in a non-denaturing PAGE gel. The two single strands of one mutated DNA typically run as two separate bands with different mobility compared to the WT bands. These single-strand DNAs (in the right gel) migrate in a much slower speed compared to double-strand DNAs (in the left gel)

Evaluation of amplicon length and mutation position for the SSCP analysis. a Schematic diagram showing positons of PCR primers and the length of amplicons. The position of deletions is indicated by red. b SSCP detection of PCR amplicons that contain 1, 2, 5 and 9-bp deletions. Single-strand DNAs were resolved in 15 % non-denaturing PAGE gels. Note the band representing double-strand DNA due to re-annealing was indicated by a red asterisk (color figure online)
SSCP can effectively and sensitively detect small indels
To test whether SSCP can be readily used to distinguish indels of any length, we made a more complete set of deletion plasmids with deletions ranging from 1 to 9 bp (Fig. 3a). These deletions were PCR amplified and evaluated by SSCP analysis. We found all PCR amplicons with different deletion sizes could be distinguished from the WT (Fig. 3b). In most cases, both denatured single-strand DNAs migrated differently from the two single strands of the WT (Fig. 3b), demonstrating that the SSCP analysis indeed has a high resolution. As a comparison, we also used PAGE to detect heteroduplex DNA for 1 and 2-bp deletions. Although 2-bp deletions were clearly detected, we could not detect 1-bp deletion with this analysis (Fig. S1). This result suggests single-strand DNA renders higher sensitivity for the detection of small indels than double-strand DNA.

Detection of a series of deletions by SSCP. a Schematic diagram showing the positon of PCR primers relative to the positions of DNA deletions. b SSCP detection of PCR amplicons that contains up to 9-bp deletions. Note the single-strand DNA was resolved on the upper position of the PAGE gel and the double-strand DNA was resolved on the lower position of the PAGE gel (indicated by a red asterisk)
In our SSCP experimental setting, we have consistently observed double-strand DNA due to partial re-annealing of single-strand DNA (Figs. 2, 3). We took advantage of this reliable phenomenon to directly compare SSCP with the detection of heteroduplex DNA in the same PAGE gel. We used PCR amplicons of 2 bp deletion and mixed it with the WT amplicons in different ratios to obtain DNA pools where mutated DNA ranged from 10 to 100 % in a 10 % gradient (Fig. 4). Such DNA mixtures were denatured and analyzed in a non-denaturing PAGE gel. We found that both single strands of DNA from the deletion amplicons migrated differently from the two single-stranded DNA of the WT amplicons (Fig. 4). The 2-bp deletion amplicons could be reliably identified when they are present as low as 10 % of total amplicon DNA (Fig. 4). Further, the intensity of single-strand DNA bands nicely reflected the DNA composition in the sample, which allows for quantification of mutation frequency. We also observed two double-strand DNA bands at the front of the PAGE gel in each lane, with the heteroduplex DNA running slightly slower than the homoduplex DNA (Fig. 4). Although heteroduplex DNA bands indicate mutations, mutation frequency could not be quantified with this method. For example, when the sample contains 100 % 2-bp deletion amplicons, the heteroduplex DNA band disappeared completely as expected, which made it indistinguishable from the WT (Fig. 4). This result demonstrates that SSCP-based detection is more reliable than heteroduplex-based detection for screening and quantifying mutations generated by CRISPR/Cas9. For example, heteroduplex-based detection methods would not be able to identify homozygous mutations which are most desirable to obtain. Since partial re-annealing of single-strand DNA always occurs in our SSCP procedure, the experiment in essence allows for simultaneous detection of single-strand DNA, heteroduplex DNA and homoduplex DNA.

Mutation frequency can be quantified by SSCP but not by the heteroduplex DNA-based method. PCR amplicon of WT was spiked with amplicons of a 2-bp deletion in a 10 % gradient. Note single-strand DNAs of WT and the mutant were resolved in the upper position of the PAGE gel. Heteroduplex and homoduplex DNAs were indicated in the lower position of the PAGE gel
Application of SSCP for detecting CRISPR/Cas9-induced targeted mutagenesis in rice
After demonstrating that SSCP could be used to sensitively detect a wide range of indels, we applied this method for mutant screen in rice with CRISPR/Cas9-induced targeted mutagenesis. We first targeted OsROC5 gene (GenBank: AB101648) in rice (Fig. 5a) (Zou et al. 2011) with an active sgRNA demonstrated previously (Feng et al. 2013; Lowder et al. 2015). Agrobacterium-mediated transformation of rice was conducted with a T-DNA construct containing Arabidopsis codon-optimized Cas9 (AteCas9) (Fauser et al. 2014) under a maize ubiquitin promoter and an OsROC5-targeting sgRNA under an OsU6 promoter. Individual T0 transgenic plants were screened by the SSCP method and many plants displayed different band profiles on the non-denaturing PAGE gels, indicating presence of mutations. Results from five such lines are shown in Fig. 5B. Based on the evidence from SSCP, #OsROC5-M02 and #OsROC5-M05 seem to contain small indel mutations. By contrast, lines #OsROC5-M01, #OsROC5-M03 and #OsROC5-M05 seem to contain large insertions based on the homoduplex DNA bands at the front of the PAGE gel (Fig. 5b). For these three lines, we also observed high molecular weight (HMW) bands at the very top of the PAGE gel. These bands could represent single-strand DNAs of large insertions or heteroduplex DNAs with large insertions or both. We cloned the PCR amplicons that span the mutations and subjected them to Sanger DNA sequencing. The three large insertions in these corresponding lines were indeed verified (Fig. 5c). In addition, we found one line is a heterozygous biallelic mutant (#OsROC5-M03) and the other four lines are heterozygous monoallelic mutants (Fig. 5c).

Application of SSCP for the detection of mutations at OsROC5 in rice induced by CRISPR/Cas9. a Schematic diagram of OsROC5 gene with the gRNA target site indicated where the PAM sequence is in red. b Detection of mutations by SSCP in five rice T0 plants. Double-strand DNAs are indicated by red asterisks. c Sequencing confirmation of mutations in each T0 lines. Note the region for SSCP is highlighted by a rectangle window and the bands in the lower position of the PAGE gel representing double-strand DNA due to re-annealing were indicated by red asterisks (color figure online)
We further tested the SSCP method at OsDEP1 (GenBank: FJ039904), an important gene that impacts grain yield in rice (Huang et al. 2009). The last exon of OsDEP1 was targeted by CRISPR/Cas9 (Fig. 6a). By screening T0 plants with the SSCP method, many promising lines were identified that show different single-strand DNA profiles on the PAGE gels. Analysis of 8 such lines is shown in Fig. 6b. We further followed these lines by Sanger sequencing. Based on the sequencing results (Fig. 7a), three lines are monoallelic mutants (#OsDEP1-m28, m31 and m35) and four lines are biallelic mutants (#OsDEP1-m32, m37, m39 and m40). One line (#OsDEP1-m30) contains two different mutations and the WT allele, indicating this plant is mosaic. With heteroduplex-based detection methods, we would not be able to identify the preferred homozygous mutant, #OsDEP1-m39, which clearly demonstrates an advantage of SSCP over other methods listed in Table 1 except HRMA. The seedling of line #OsDEP-32 was transplanted to soil along with a WT control plant. We found this plant indeed displayed a reduced length of the inflorescence internode, an anticipated phenotype when OsDEP1 is knocked out (Huang et al. 2009).

Application of SSCP for the detection of mutations at OsDEP1 in rice induced by CRISPR/Cas9. a Schematic diagram of OsDEP1 gene with the gRNA target site indicated where the PAM sequence is in red. b Detection of mutations by SSCP in multiple T0 plants. Homoduplex DNA at the very front of the gel is indicated by a red asterisk. C. Detection of mutations by the formation of heteroduplex DNA. Homoduplex DNA at the very front of the gel is indicated by a red asterisk (color figure online)

Genotype and phenotype of OsDEP1 mutants. a Sequencing confirmation of mutations in each T0 lines. b Phenotype of OsDep1 #32 rice line which is a biallelic heterozygous mutant. It shows a dwarf stature of the mutant in comparison to the WT
In this SSCP analysis, we again found multiple HMW bands in #OsDEP1-m30 and #OsDEP1-m40 lines (Fig. 6b). Based on our previous observation (Fig. 5b), such HMW bands suggested large insertions, which were not confirmed by subsequent DNA sequencing in this case (Fig. 7a). Rather, we identified large deletions of 39 bp in both samples. To understand the nature of these HMW bands, we analyzed the same 8 lines using the method based on detection of heteroduplex DNA in a PAGE gel (Ota et al. 2013; Zhu et al. 2014). Interestingly, HMW bands were also observed in #OsDEP1-m30 and #OsDEP1-m40 lines in this experiment (Fig. 6c), which strongly suggests the presence of large deletions that could result in multiple forms of heteroduplex DNA that have poor mobility in a PAGE gel. Hence, we concluded that HMW bands in #OsDEP1-m30 and #OsDEP1-m40 lines were re-annealed heteroduplex DNA with one strand containing a large deletion. Such heteroduplex DNA forms with high sequence heterogeneity would result in a large singe-strand DNA bulge, which could significantly impact the mobility during electrophoresis.
Discussion
Besides direct Sanger DNA sequencing, four major types of methods have been used for the detection of mutations generated by SSNs such as ZFN, TALEN and CRISPR/Cas9 (Table 1). The first method is based on restriction fragment length polymorphism (RFLP), in which SSN-induced mutations coincidently abolish the recognition sequence of a restriction enzyme (Urnov et al. 2005). We have used this reliable methods for assaying targeted mutagenesis with ZFN (Qi et al. 2013a, b, 2014), TALEN (Christian et al. 2013; Zhang et al. 2013) and CRISPR/Cas9 (Lowder et al. 2015). One large drawback of this method is that the selection of target sites is limited by the availability of useful restriction enzyme sites. Further, different restriction enzymes are often required for different target sites and it thus requires a large collection of restriction enzymes if SSN-mediated targeted mutagenesis is routinely pursued in a lab. The three other methods are all based on the formation of heteroduplex of DNA (Table 1). In a Surveyor assay (Cel1) (Miller et al. 2007; Oleykowski et al. 1998) or a T7 endonuclease assay (T7E1) (Mashal et al. 1995), such heteroduplex DNA is cleaved by the enzyme and subsequently resolved in an agarose and PAGE gel. Compared to the restriction enzyme-based RFLP method, Surveyor or T7 endonuclease assay could be more versatile because it does not restrict target choice of SSNs or require many enzymes (Vouillot et al. 2015). However, Surveyor or T7 assay is usually not as reliable as the restriction enzyme-based RFLP method during experiments. In high-resolution melting analysis (HRMA) (Dahlem et al. 2012), heteroduplex and homoduplex DNA is labeled with DNA dye and detected by specific instruments, such as quantitative PCR machines. HRMA has a high resolution and throughput for distinguishing amplicons with different SNPs, but it requires quantitative PCR machines which may not be readily available in every lab. Finally, PAGE-based assay is another way to detect heteroduplex DNA (Ota et al. 2013; Zhu et al. 2014), which runs slower than homoduplex DNA, such as WT DNA. Due to its intrinsic limitation, all of these heteroduplex DNA-based methods cannot identify homozygous mutants, which in fact are the most desired outcomes of targeted mutagenesis with SSNs.
Distinct from the four above-mentioned methods, the SSCP method is based on the detection of single-strand DNA (Fig. 1; Table 1). In this assay, PCR amplicons are denatured and run as single-strand DNA forms in a non-denaturing PAGE gel. To achieve a high-resolution separation in a PAGE gel, we found PCR amplicons need to be smaller than 300 bp since single-strand DNA moves much slower than double-strand DNA during electrophoresis (Fig. 2). We have shown that it is relatively sensitive to detect small indel mutations (Figs. 2, 3). For example, it can reliably distinguish 1 bp indels, while the PAGE-based assay for heteroduplex DNA sometimes failed to detect such mutations (Fig. S1). CRISPR/Cas9 generates a high proportion of 1 bp indels in plants (Feng et al. 2014; Ma et al. 2015; Zhang et al. 2014) and likely in other species as well. More importantly, SSCP is capable of identifying homozygous mutants (Figs. 6, 7). Thus, SSCP enables the detection of a wide range of mutations without missing homozygous mutants.
In the SSCP assay, it is difficult to prevent the re-annealing of a fraction of single-stand DNA in the samples (Figs. 2b, 3b, 4, 5b, Fig. 6b). However, the resulting double-strand DNA (either homoduplex or heteroduplex) can be easily distinguished from single-strand DNA due to the drastic difference in mobility during electrophoresis (Figs. 2b, 3b, 4b), and hence does not affect the overall analysis. The intrinsic nature of single-strand DNAs causes their migration pattern in a non-denaturing PAGE gel to be affected by size as well as sequence. Thus, it is sometime difficult to tell whether a differentially migrated band represents a deletion, insertion or base-pair change. We reason this limitation is not very important because the nature of mutations will be identified through Sanger DNA sequencing in the next step. In a mutant screen experiment, the most important thing is to first identify individuals that carry mutations, particularly those that carry homozygous biallelic mutations. As we demonstrated, the SSCP analysis allows for simultaneous detection of both single-strand and double-strand DNAs resulting in accurate identification of homozygous biallelic mutants. Hence, this method is more powerful than the methods solely based on the detection of heteroduplex DNA by PAGE (Ota et al. 2013; Zhu et al. 2014).
In conclusion, we found that the SSCP analysis is a reliable method for detection of a wide range of indels or mismatch mutations with CRISPR/Cas9. Importantly, this method can identify the most desirable homozygous mutants, which could be missed by other popular heteroduplex-based methods (Table 1). We have successfully applied the SSCP method to screen transgenic rice T0 plants for mutant identification (Figs. 5, 6, 7). Targeted mutagenesis by CRISPR/Ca9 generally occurs at high frequencies in rice, which seemingly makes prescreen methods such as SSCP (Table 1) less useful because Sanger sequencing can be directly pursued. However, we believe these prescreen methods will continue to be used in many plant species and other organisms where genome editing frequency is relatively low.
Author contribution statement
YZ and YQ conceived and designed the experiments. XZ, SY, DZ, ZZ and XT performed the experiments. YZ, YQ, XZ, SY, DZ, KD and JZ analyzed the data. YZ, YQ and XZ wrote the paper.
References
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188:773–782
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761
Christian M, Qi Y, Zhang Y, Voytas DF (2013) Targeted mutagenesis of Arabidopsis thaliana using engineered TAL effector nucleases. G3 3:1697–1705
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ (2012) Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet 8:e1002861
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
Endo M, Mikami M, Toki S (2015) Multigene knockout utilizing off-target mutations of the CRISPR/Cas9 system in rice. Plant Cell Physiol 56:41–47
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM (2013) Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10:1116–1121
Fauser F, Schiml S, Puchta H (2014) Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant J 79:348–359
Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, Zhu JK (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang DL, Wang Z, Zhang Z, Zheng R, Yang L, Zeng L, Liu X, Zhu JK (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA 111:4632–4637
Gaj T, Gersbach CA, Barbas CF 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282
Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA (2013) Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110:15644–15649
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X (2009) Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41:494–497
Ikeda T, Tanaka W, Mikami M, Endo M, Hirano HY (2015) Generation of artificial drooping leaf mutants by CRISPR-Cas9 technology in rice. Genes Genet Syst 90:231–235
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41:e188
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Kakavas VK, Plageras P, Vlachos TA, Papaioannou A, Noulas VA (2008) PCR-SSCP: a method for the molecular analysis of genetic diseases. Mol Biotechnol 38:155–163
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93:1156–1160
Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B (2011) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39:359–372
Lowder LG, Zhang D, Baltes NJ, Paul JW, Tang X, Zheng X, Voytas DF, Hsieh TF, Zhang Y, Qi Y (2015) A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol 169:1–15
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, Wang B, Yang Z, Li H, Lin Y, Xie Y, Shen R, Chen S, Wang Z, Chen Y, Guo J, Chen L, Zhao X, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Mashal RD, Koontz J, Sklar J (1995) Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat Genet 9:177–183
Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res 23:1233–1236
Mikami M, Toki S, Endo M (2015) Comparison of CRISPR/Cas9 expression constructs for efficient targeted mutagenesis in rice. Plant Mol Biol 88:561–572
Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148
Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT (1998) Mutation detection using a novel plant endonuclease. Nucleic Acids Res 26:4597–4602
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T (1989a) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 86:2766–2770
Orita M, Suzuki Y, Sekiya T, Hayashi K (1989b) Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5:874–879
Ota S, Hisano Y, Muraki M, Hoshijima K, Dahlem TJ, Grunwald DJ, Okada Y, Kawahara A (2013) Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells: Devot Mol Cell Mech 18:450–458
Paques F, Duchateau P (2007) Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther 7:49–66
Qi Y, Li X, Zhang Y, Starker CG, Baltes NJ, Zhang F, Sander JD, Reyon D, Joung JK, Voytas DF (2013a) Targeted deletion and inversion of tandemly arrayed genes in Arabidopsis thaliana using zinc finger nucleases. G3 3:1707–1715
Qi Y, Zhang Y, Zhang F, Baller JA, Cleland SC, Ryu Y, Starker CG, Voytas DF (2013b) Increasing frequencies of site-specific mutagenesis and gene targeting in Arabidopsis by manipulating DNA repair pathways. Genome Res 23:547–554
Qi Y, Starker CG, Zhang F, Baltes NJ, Voytas DF (2014) Tailor-made mutations in Arabidopsis using zinc finger nucleases. Methods Mol Biol 1062:193–209
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186–191
Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355
Sekiya T (1996) Single-strand conformation polymorphism (SSCP) analysis: a convenient, rapid method for detection of single-base changes in DNA. Tanpakushitsu kakusan koso Protein, nucleic acid, enzyme 41:539–545
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31:686–688
Shirasawa K, Monna L, Kishitani S, Nishio T (2004) Single nucleotide polymorphisms in randomly selected genes among japonica rice (Oryza sativa L.) varieties identified by PCR-RF-SSCP. DNA Res: Int J Rapid Publ Rep Genes Genomes 11:275–283
Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, Montoya G, Paques F, Duchateau P (2006) A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res 34:e149
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67
Stewart CN Jr, Via LE (1993) A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques 14:748–750
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11:636–646
Vouillot L, Thelie A, Pollet N (2015) Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 5:407–415
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112:3570–3575
Xu RF, Li H, Qin RY, Li J, Qiu CH, Yang YC, Ma H, Li L, Wei PC, Yang JB (2015) Generation of inheritable and “transgene clean” targeted genome-modified rice in later generations using the CRISPR/Cas9 system. Sci Rep 5:11491
Zhang Y, Zhang F, Li X, Baller JA, Qi Y, Starker CG, Bogdanove AJ, Voytas DF (2013) Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol 161:20–27
Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, Zhu JK (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12:797–807
Zhou H, Liu B, Weeks DP, Spalding MH, Yang B (2014) Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res 42:10903–10914
Zhu X, Xu Y, Yu S, Lu L, Ding M, Cheng J, Song G, Gao X, Yao L, Fan D, Meng S, Zhang X, Hu S, Tian Y (2014) An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci Rep 4:6420
Zou LP, Sun XH, Zhang ZG, Liu P, Wu JX, Tian CJ, Qiu JL, Lu TG (2011) Leaf rolling controlled by the homeodomain leucine zipper class IV gene Roc5 in rice. Plant Physiol 156:1589–1602
Acknowledgments
The pBlueScript-derived constructs were kind gifts from Satoshi Ota and Atsuo Kawahara at RIKEN Institute in Japan. This work is supported by Grants including the National Science Foundation of China (31330017, 31271420 and 31371682), the national Transgenic Major Project (2014ZX0801003B-002) and the Fundamental Research Funds for the Central Universities (ZYGX2013J099) to YZ, and startup funds from East Carolina University and a Collaborative Funding Grant (2016-CFG-8003) from North Carolina Biotechnology Center and Syngenta to YQ.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict interests.
Additional information
Communicated by T. Cardi.
X. Zheng, S. Yang and D. Zhang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
299_2016_1967_MOESM2_ESM.pptx
Supplementary material 2 (PPTX 71 kb) Fig. S1 PAGE-based detection of heteroduplex DNA of 1-bp and 2-bp deletions. This method is not as sensitive as SSCP because it failed to detect the 1-bp deletion in this case
Rights and permissions
About this article

Cite this article
Zheng, X., Yang, S., Zhang, D. et al. Effective screen of CRISPR/Cas9-induced mutants in rice by single-strand conformation polymorphism. Plant Cell Rep 35, 1545–1554 (2016). https://doi.org/10.1007/s00299-016-1967-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-016-1967-1