
Abstract
Key message
We report success of host-induced gene silencing in downregulation of aflatoxin biosynthesis in Aspergillus flavus infecting maize transformed with a hairpin construct targeting transcription factor aflR.
Abstract
Infestation of crops by aflatoxin-producing fungi results in economic losses as well as negative human and animal health effects. Currently, the control strategies against aflatoxin accumulation are not effective to the small holder farming systems in Africa and this has led to widespread aflatoxin exposure especially in rural populations of sub-Saharan Africa that rely on maize as a staple food crop. A recent strategy called host-induced gene silencing holds great potential for developing aflatoxin-resistant plant germplasm for the African context where farmers are unable to make further investments other than access to the germplasm. We transformed maize with a hairpin construct targeting the aflatoxin biosynthesis transcription factor aflR. The developed transgenic maize were challenged with an aflatoxigenic Aspergillus flavus strain from Eastern Kenya, a region endemic to aflatoxin outbreaks. Our results indicated that aflR was downregulated in A. flavus colonizing transgenic maize. Further, maize kernels from transgenic plants accumulated significantly lower levels of aflatoxins (14-fold) than those from wild type plants. Interestingly, we observed that our silencing cassette caused stunting and reduced kernel placement in the transgenic maize. This could have been due to “off-target” silencing of unintended genes in transformed plants by aflR siRNAs. Overall, this work indicates that host-induced gene silencing has potential in developing aflatoxin-resistant germplasm.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Contamination of food crops by aflatoxins is a serious food safety concern in sub-Saharan Africa (Lewis et al. 2005; Mutegi et al. 2009). Recently, targeted downregulation of gene expression by small interfering RNAs (siRNAs) has been successful in engineering crops against viruses (Duan et al. 2012), insects (Huvenne and Smagghe 2010), nematodes (Niu et al. 2006), parasitic plants (Alakonya et al. 2012) and fungi (Nunes and Dean 2012). These biotechnological breakthroughs have opened up a platform for application of this technique in developing resistant crops against other stresses that have not been reported such as aflatoxins (Alakonya and Monda 2013). Although host-induced gene silencing (HIGS) resulting from transfer of siRNAs from plants to fungi has been demonstrated many times (Voegele and Mendgen 2003; Tinoco et al. 2010; Nowara et al. 2010; Micali et al. 2011; Yin et al. 2011; Koch et al. 2013), the mechanism remains unclear, but fungal vesicles and plant-fungal cellular interface have been proposed as possible trafficking channels (Koch et al. 2013: Micali et al. 2011; Voegele and Mendgen 2003). Since elucidation of the aflatoxin biosynthetic pathway, greater understanding of pathway intermediates, enzymes involved and mechanisms of regulation has been achieved (Bennet and Klich 2003; Yu et al. 2004). This pathway involves 30 genes clustered on a 70 Kb region of chromosome III of the fungal genome and has a DNA-binding protein (AflR) that is encoded by an aflatoxin regulatory transcription factor (aflR). The aflR transcription factor is a key regulator of most structural genes in the aflatoxin biosynthetic pathway (Amaike and Keller 2011; Yu 2012). In fact, disruption of this transcription factor thwarts expression of genes in the aflatoxin biosynthetic pathway (Payne et al. 1993; Cary et al. 2000). This understanding therefore formed the basis for our targeted downregulation of the aflR transcription factor through host-induced gene silencing.
HIGS is achieved via expression of double-stranded (ds) RNAs into plants through transient or stable transformation protocols using RNAi vectors (Wesley et al. 2001). This type of post-transcriptional gene silencing (PTGS), termed co-suppression in plants and quelling in fungi, is mediated by 21–24 nt dsRNA molecules known as siRNAs produced by an endonuclease called Dicer (Hamilton and Baulcombe 1999). The siRNAs are incorporated in an RNA-induced Silencing Complex (RISC) that then guides them to degrade complementary target mRNAs in a sequence specific manner (Baulcombe 2004). In our strategy, we transformed maize with an RNAi construct targeting 778 bp of the aflR sequence. We reasoned that the construct would generate primary siRNAs in the host plant and upon infection by aflatoxigenic fungi, the primary siRNAs would cross into the fungi and cleave complementary aflR mRNA sequences hence thwarting the expression of other pathway genes and resulting in downregulation of aflatoxin biosynthesis.
Materials and methods
Construct preparation
The RNAi sequence used in this study was cloned from a highly aflatoxigenic A. flavus isolate from Machakos, an area endemic to aflatoxin outbreaks in Kenya. This isolate, also used for infection assays, will be referred to as MCKII throughout the text. For PCR, aflR specific primers (Table 1) were designed based on the NCBI gene bank sequence (accession number gi/19908787/gb/AF441439.1) and used to amplify the aflR gene from cDNA synthesized from A. flavus. The PCR product was first cloned into PCR8/GW/TOPOTA entry vector (Invitrogen Corp. Carlsbad CA, USA), and then transferred into binary vector pStargate (with Ubiquitin promoter) through the recombination process of the Gateway cloning technology (Invitrogen Corp. Carlsbad CA, USA). Recombination results into insertion of two aflR gene fragments between a promoter and a terminator sequence in sense and antisense orientations separated by a pdk intron. This construct is indicated hp-aflR throughout the text (Fig. 1). Verification for presence of the construct in the binary vector was done through restriction digestion using XhoI (New England Bio-labs Inc., MA, USA).

A map showing the T-DNA region of binary vector pStargate with the aflR sequences in sense and antisense orientations. From the RB-right border is the HPTII-hygromycin phosphotransferase resistance gene for plant selection driven by the 35S cauliflower mosaic virus promoter and terminator at the downstream (not shown) of the plant selection marker. The silencing sequences are driven by the maize ubiquitin promoter while aflR sense and antisense sequences are separated by pdk intron with an octopine synthase terminator (OCS) downstream
Production of transgenic plants
The RNAi construct was introduced into Agrobacterium tumefaciens, strain EHA 101, and then used for transformation of maize tropical inbred line CML 144 (CIMMYT). Maize transformation was done according to Ishida et al. (2007). Selection of putatively transformed maize calli was achieved on Murashige and Skoog (MS) medium (Murashige and Skoog 1962) supplemented with hygromycin. Acclimatization and hardening of putative transgenic plantlets was done on peat moss in the glasshouse.
Confirmation of transgenic plants
Putatively transformed plants were screened for presence of the silencing construct via regular PCR. First, genomic DNA from T0 plants was isolated as described by Zidani et al. (2005). Hygromycin specific primers (Table 1) were then used for amplification in 20 µL reaction mix (containing 10 ng genomic DNA, 1X KAPA Taq and 1 µM of each primer) using a DNA thermocycler (Eppendorf AG, Hamburg, Germany) at an annealing temperature of 61 °C. Expression of the construct in transgenic maize (T1) was verified through reverse transcriptase polymerase chain reaction (RT-PCR) analysis using hygromycin (plant selectable marker) specific primers with the 18S rRNA specific primers as an internal control for maize (Table 1). Briefly, total RNA was isolated from leaf tissues of each of the first generation (T1) plants using the RNeasy Plant Mini kit (Qiagen) including a step for on column DNase digestion. The RNA was quantified using a nanodrop and 1 µg reverse transcribed using First strand Superscript III reverse transcriptase kit (Invitrogen Corp. Carlsbad CA, USA) using random heximers followed by treatment with RNase H to degrade any remaining RNA. PCR amplification was performed in 20 µL reaction mix (containing 10 ng cDNA, 1X KAPA Taq and 1 µM of each primer) using a DNA thermocycler (Eppendorf AG, Hamburg, Germany) at an annealing temperature of 61 °C for both primer sets.
Molecular determination of silencing of target aflR gene in aflatoxigenic A. flavus
To investigate silencing of target aflR transcripts in A. flavus re-isolated from transgenic maize harboring the hp-aflR construct, we carried out an RT-PCR analysis. RNA was isolated from 5 days’ old A. flavus cultures grown on yeast extract sucrose (YES) media. Approximately 200 mg of mycelia were recovered with a sterile spatula, dried in absorbent paper and ground with liquid nitrogen in a sterile cold mortar and pestle. Total RNA was extracted using the RNeasy Plant Mini Kit with on column DNase digestion (Qiagen) according to manufacturer’s instructions. One microgram of RNA was then subjected to cDNA synthesis using Superscript III reverse transcriptase kit (Invitrogen). RT-PCR was performed in 20 μL reactions comprising 8 μL of One-Step RT-PCR Pre-Mix kit, 0.2 μM of each primer and 1 μL of template cDNA. Gene specific primers (aflR) were designed with the reverse primer, labeled aflR-R for RT-PCR in Table 1, annealing outside the cloned region. Tubulin primers were also included as an internal amplification control for fungi.
Aflatoxin analysis
To determine if transgenic maize expressing hp-aflR siRNAs inhibit aflatoxin production and accumulation, we challenged first generation transgenic maize (T1) with aflatoxigenic A. flavus. First, we used a non-wounding technique for maize infection (Windham and Williams 2007) under controlled conditions in the glasshouse. Glasshouse conditions were 12 h light/12 h darkness photoperiod, with temperatures maintained between 26 and 28 °C and relative humidity of 60 %. Transgenic maize plants maintained in the glasshouse were infected with the toxigenic A. flavus culture through the silk to mimic what happens in nature. Infected silks were then covered with a clean plastic bag for 3 days to ensure a moist environment for fungal colonization. Ten (10) plants each from each of the 5 independent transformation events were used in the aflatoxin assays with 5 non-transformed CML 144 plants also included as controls. The plants were watered regularly until maturity upon which seeds were collected for further analysis. Upon collection of seeds, A. flavus colonizing the maize kernels was re-isolated using the direct plating technique on potato dextrose agar medium as earlier described by Pitt and Hockings (1997). The pure fungal colonies were subsequently used to test aflatoxigenicity.
Toxigenic potential of the re-isolated A. flavus was first determined by inoculating pure fungal cultures on neutral red desiccated coconut agar (NRDCA) medium and visualized under UV light as described by Atanda et al. (2006). To determine the levels of aflatoxins accumulating in infected transgenic and wild type maize kernels, we carried out an enzyme-linked immunosorbent assay (ELISA) on the collected seeds. Ten kernels from each of the five independent events were subjected to ELISA using the RIDASCREEN® extraction kit (R-Biopharm AG, Darmstadt, Germany) according to the manufacturer’s instructions. Extracted aflatoxins were then quantified by measuring absorbance of the samples using a microtiter spectrophotometer at 450 nm. Further, A. flavus cultures re-isolated from transgenic and wild type maize above were also analyzed for aflatoxin production using a protocol adopted from Bragulat et al. (2001).
Measurement of plant growth parameters
To investigate the effect of hp-aflR on plant growth, first generation transgenic maize plants were maintained in the glass house and measurements on various growth parameters taken. Data on growth parameters were collected on five transgenic maize plants randomly sampled from five independent transformation events. Five non-transformed CML 144 plants were also included as controls. Plant heights were taken upon silking using a tape measure and their respective averages calculated. For yield determination, the number of kernels per cob was counted after harvest and their averages calculated. Wet weights of the cobs were then taken using a weight balance and recorded before incubating them at 65 °C in an oven for 3 days to get rid of excess moisture. Upon drying, dry weights of the cobs from transgenic and wild type plants were determined using the weight balance. Analysis of variance (ANOVA) on the plant height and weights was performed using SAS version 9.1 and comparisons drawn.
Bioinformatics analysis
To identify aflR homologs in maize and groundnuts, BLASTX searches were performed on non-redundant (NR) databases of nucleotide sequences at National Center for Biotechnology Information (NCBI, National Institutes of Health, Bethesda, MD) (http://www.ncbi.nlm.nih.gov/BLAST) and at Plant Transcription Factors Database (PTFD) (Center for Bioinformatics, Perking University, China) (http://www.planttfd.cbi.pku.edu.cn) with an E value cutoff of 1−e10. The aflR sequence from A. flavus used in cloning above was retrieved from available genomic sequences at NCBI. The aflR domain sequence was scored against NR maize transcription factor database using the PFAM matrix. Hits with PFAM trusted cutoffs were retrieved and considered for further analysis. Multiple sequence alignment (MSA) was carried out using Vector NTI Advance™ version 10. The aflR transcript sequence was also used as a reference to query and predict potential off-targets between its siRNAs and the two ERFs identified after BLAST search on PTFD on the Si-Fi software v3.1 platform (http://labtools.ipk-gatersleben.de). The parameters used included a 5′ antisense strand, starting with an A or U base, a 5′ sense strand, starting with G or C, and the first seven bases from a 5′ antisense strand end containing at least three to five A/U bases.
Results
Transformation of maize with hp-aflR
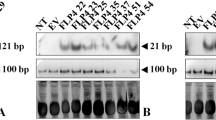
Successful preparation of the hp-aflR construct was achieved following a perfect pairwise alignment between a 778 bp aflR fragment and the full cDNA sequence retrieved from the gene bank as shown in the figure (Online Resource 1). Maize embryos were successfully transformed with the hp-aflR construct (Fig. 1) and regenerated using a protocol by Ishida et al. (2007). Selection of putative hairpin aflR transgenic maize plants was achieved using hygromycin with selected calli taken through the regeneration procedure until hardening and acclimatization on peat moss (Fig. 2a–e). A relatively high frequency of transformation was achieved for the inbred line CML 144 resulting in several independent transgenic events (Table 2). PCR amplification on genomic DNA revealed the expected 861 bp fragment of the hygromycin selectable marker gene in T0 plants (Fig. 2f). This confirmed the transgenic status of these plants. RT-PCR analysis of first generation maize plants transformed with the hp-aflR transgene revealed that 28 out of the 41 putative transformants were transgenic, producing the expected 861 bp fragment of the hygromycin gene. The hygromycin gene expression was not detected in wild type (Fig. 3a).

Stages in transformation and regeneration of putative transgenic maize plants. a Putatively transformed maize calli maintained on MS medium with 30 mg/l hygromycin for selection. b Compact callus obtained after second selection showing presence of somatic embryos. c Multiple maize shoots on regeneration medium. d Acclimatization of a maize plantlet on peat moss in the glass house. e T0 (aflR) plants in the glass house alongside non-transformed (WT) maize. f PCR amplification of the hygromycin selectable marker gene in T0 maize plants. M represents a 50 bp DNA ladder, −ve is the negative PCR amplification control, WT is amplification of genomic DNA from non-transformed maize while aflR 1, 2 and 3 represent amplification in plants randomly sampled from independent transgenic events

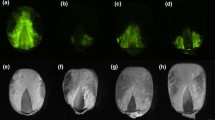
Determination of effect of introduction of the hp-aflR construct in maize on aflR gene expression and aflatoxin production in A. flavus. a Expression of hp-aflR construct in transgenic maize through RT-PCR. The expected fragment (861 bp) of the hyromycin selectable marker gene was amplified on cDNA prepared from three putatively transformed maize events (aflR) but not in the non-transformed (WT) plant alongside 18S rRNA used as internal control for maize. b Silencing of the aflR gene in A. flavus cultures colonizing transgenic and non-transformed (WT) maize. The intensity of the bands reduced in A. flavus cultures colonizing transgenic maize as compared to those infecting non-transformed maize (WT) and the positive control (MCKII) not infecting any maize. Amplification of the β-tubulin rRNA was used as internal control for fungus. c Screening for in vitro production of aflatoxin on neutral red desiccated coconut agar media indicating low fluorescence intensity in A. flavus colonizing transgenic maize (aflR) when compared with fungal cultures infecting the wild type (WT) and that not infecting any maize (MCKII). M represents a 1 Kb DNA molecular marker, −ve is a negative control during PCR amplification, WT is non-transformed maize while aflR 1, 2 and 3 represent independent transgenic events with hp-aflR silencing construct
Expression of hp-aflR transgene in maize downregulates aflR expression in infecting A. flavus and reduces aflatoxin accumulation in the host plant
To assess whether expression of the hp-aflR construct reduces aflatoxin accumulation in maize, we infected the silks of transformed and non-transformed maize plants with aflatoxigenic A. flavus 6 days after pollination. RT-PCR results showed reduced signals in A. flavus cultures recovered from transgenic maize as compared to those from wild type plants and the culture used for infection (Fig. 3b). Furthermore, MCKII fungi used for infection and that re-isolated from wild type maize showed a bright blue fluorescence on neutral red desiccated coconut agar media characteristic of high aflatoxigenicity (Atanda et al. 2011). Moderately bright fluorescence was observed in fungal cultures re-isolated from maize transformed with hp-aflR construct (Fig. 3c). Similarly, ELISA results revealed that transgenic maize kernels accumulated an average of 10 µg/kg total aflatoxins that is just at the threshold limit permitted in foodstuffs by the Kenya Bureau of Standards (KEBS) (Fig. 4a). These aflatoxin levels were significantly lower compared with those obtained from the wild type maize (141 µg/kg), also infected with the A. flavus isolate, according to Tukey’s test (P < 0.05). Furthermore, A. flavus cultures re-isolated from wild type maize recorded a higher level of total aflatoxins (117 µg/kg) than the fungal culture used for infection (MCKII) which produced 70.3 µg/kg and those from transgenic maize (aflR) at 35.9 and 43.6 µg/kg (Fig. 4b).

Levels of total aflatoxins. a Total aflatoxins accumulated in non-transformed maize (WT) and transgenic (aflR) maize transformed with an hp-aflR construct. The red line indicates the maximum limit of total aflatoxins in foodstuffs permitted by the Kenya Bureau of Standards (KEBS). b Total aflatoxins in A. flavus cultures re-isolated from non-transformed maize (WT) and from maize with hp-aflR construct (aflR Rep 1 and aflR Rep 2) alongside the culture used for infection (MCKII). Values on the Y axis are an average spectrophotometer readings for five randomly selected maize samples from five independent transformation events. Error bars indicate the standard errors of the mean. Treatment means with the same letter are not significantly different according to Tukey’s HSD test (P < 0.05, n = 5)
Expression of the hp-aflR transgene negatively affects growth and yield of maize
To investigate the possible effects of the hp-aflR transgene on growth and yield of transgenic maize, we measured the plant height, cob weight and the number of kernels per cob. We found out that there was a significant decrease in plant height and cob weight in transgenic maize plants in comparison to the wild type (Fig. 5a–d). Generally, transgenic maize exhibited small cobs that were poorly filled (Fig. 5d). On average, transgenic maize recorded a height of 100 cm that represents a significant 2.5-fold reduction in relation to the corresponding wild type plants (Fig. 5e). A reduction in the weight of cobs at harvest was also observed in transgenic maize when compared with the wild type. The five independent transgenic events analyzed exhibited a large decrease in wet weights of the cobs (8.5-fold) lower than the wild type. As expected, a similar significant decrease was recorded after drying the cobs to a moisture content of about 13 % (Fig. 5f). The hp-aflR transgenic maize plants produced cobs that exhibited relatively lower number of kernels per cob than the corresponding wild types. Analyzed cobs from the five independent transgenic events recorded an average of 90 kernels per cob as compared to 315 kernels per cob in the non-transformed plants, which represents a significant reduction in kernel number (Fig. 5g). Interestingly, we observed that some of the cobs from transgenic events had no kernels at all even after self pollination.

Determination of the effect of introduction of hp-aflR transgene on growth and yield in maize. a A representative image of transgenic plants (aflR) with controls (WT) at silking. Images of maturing non-transformed (b) and transgenic (c) cobs after pollination. d Cob filling in transgenic (aflR) and non-transformed (WT) plants. Some cobs from transgenic events recorded empty cobs. e The average plant height reduced in transgenics expressing hp-aflR transgene (aflR) as compared to the corresponding non-transformed controls (WT). f Average dry weight of transgenic maize cobs alongside the wild types. g Mean number of kernels per cob in transgenic (aflR) alongside wild type (WT) plants. Error bars indicate the standard errors of the mean. Means with same letter are not significantly different according to Tukey’s HSD test (P < 0.05, n = 5)
BLAST analysis
To identify any aflR homologs present in maize that could have been “accidentally” targeted for silencing by the hp-aflR construct at the mRNA level, we performed a series of BLAST searches using the aflR sequence at NCBI and at the plant transcription factor database (PTFD). Our searches for aflR homologous sequences in other organisms at NCBI revealed a number of sequences, mostly from Aspergillus spp. No homologous sequences could however be identified in plants. BLAST results at the PTFD did not reveal any hits using the recommended cutoff value (1−e10). Raising the cutoff value, however, revealed two hits of ethylene responsive factors (ERF) transcription factors in maize. The obtained ERFs represent a bZIP family protein (accession number GRMZM2G136266_P01) and a GATA family protein (accession number GRMZM2G464037_P01) and show sequence similarities of 31 and 21 %, respectively to the aflR query.
The two coding sequences are involved in diverse functions in cellular processes in maize, such as DNA-dependent regulation of transcription as well as regulation of sequence-specific DNA-binding activity. They are also involved in control of various essential stages including shoot apex development, tassel and ear inflorescence and pollen development stages. To investigate presence of conserved motifs between the retrieved sequences and the aflR transcription factor, we mapped nucleotide sequences of the ERFs to the aflR query for a multiple sequence alignment using Vector NTI Advance™ and found no perfect alignment. A further analysis with Si-Fi tool did not reveal any potential off-targets between aflR siRNAs and the two ERFs.
Discussion
Host-induced gene silencing has, over the last few years, been successfully used to control various stresses caused by fungi in plants (Nunes and Dean 2012). The recent proofs of concept for HIGS through expression of hairpin cassettes in plants for control of pathogenic Fusarium verticilloides (Tinoco et al. 2010), mycotoxin-producing Fusarium graminearum (Koch et al. 2013), leaf rust fungus Puccina striiformis (Yin et al. 2011) and powdery mildew fungus Blumeria graminis (Nowara et al. 2010) opened up a platform for the use of this mechanism in controlling phytopathogenic fungi such as aflatoxin-producing A. flavus. Here, we showed that hairpin aflR cassette transformed into tropical maize line CML 144 not only silenced aflR transcripts in A. flavus cultures colonizing transgenic maize but also reduced aflatoxin biosynthesis and accumulation in infected kernels. To the best of our knowledge, this is the first in planta report demonstrating targeted aflatoxin downregulation in a food crop.
In the past, reports have shown success in disruption of aflatoxin biosynthesis in vitro through knockdown/knockout techniques targeting various genes that control rate-limiting steps in the pathway (Brown et al. 1996; Magan et al. 2011). The aflR transcription factor, which has also been targeted in previous experiments for in vitro disruption of aflatoxin biosynthesis, is a positive acting regulatory factor that is involved in transcription activation of structural genes in the aflatoxin biosynthetic pathway (Cary et al. 2000; Yu 2012). Moreover, previous experiments have implicated aflR inverted repeat transgenes in aflatoxin suppression in in vitro cultured A. flavus and A. parasiticus (McDonald et al. 2005) making it an ideal candidate for HIGS to control biosynthesis of the mycotoxin and its accumulation in plants.
The effect of hp-aflR on aflatoxin biosynthesis and accumulation in maize was notable. A possible explanation for this is that transgenic maize synthesized siRNAs that were complementary to the aflR transcription factor in A. flavus infecting the maize. Since A. flavus is a necrotrophic fungus, we postulate that upon colonization of transgenic plants, host derived siRNA molecules trafficked into infecting fungi, possibly through vesicles (Koch et al. 2013). On reaching the fungus, siRNA molecules that were complementary to the aflR sequence were recruited into the silencing machinery. The newly recruited siRNAs then chopped the aflR transcripts into 21–28 bp fragments in a fashion referred to as quelling leading to lack/low level activation of genes that act downstream of the aflatoxin biosynthesis pathway in A. flavus (Hammond et al. 2001). Low levels of functional aflR mRNAs, as revealed by our RT-PCR data, disrupted the full transcription of pathway genes and subsequently interrupted aflatoxin biosynthesis. These results support those by Magan et al. (2011) who demonstrated that aflatoxin production is disrupted if any step in the biosynthetic pathway is blocked. This work provides a proof of concept that aflatoxin biosynthesis can indeed be downregulated in planta through silencing of genes controlling the biosynthetic pathway as earlier proposed by (Alakonya and Monda 2013).
Detrimental effects to plant physiology observed in maize transformed with the RNAi cassette are a pitfall to the use of HIGS for controlling aflatoxins in plants by targeting aflR. Effects on essential physiological processes including plant elongation, synchronized silking and tassel formation as well as kernel setting and their placement on the maize cobs point towards a possible off-target effect of the transgene on the plants. Off-target silencing effects occur when siRNAs are processed by RISC and downregulate unintended target mRNAs (Xu et al. 2006). Such targeting has been shown to lead to changes in development in targeted organisms with effects on the phenotype in both plants and animals (Jackson et al. 2003; Xu et al. 2006). Reports have also demonstrated that there exists a high risk of off-target gene silencing during PTGS in plants due to a cross reaction of siRNAs with targets of non-specific nature or those of limited sequence similarity. Theoretically, siRNAs derived from cleavage of dsRNAs by Dicer are responsible for high efficiency RNA silencing of target sequences in host plants. However, studies have also implicated these short nucleotides to the suppression of unintended genes containing identical or mismatched sequences to the siRNAs (Jackson et al. 2003; Jackson and Linsley 2004; Saxena and Jonsson 2003; Snove and Holen 2004; Xu et al. 2006).
The negative effect on the maize is not the reason for the low aflatoxin levels in transgenic maize but HIGS. We reason that the alteration in plant architecture could have been due to siRNA mis-targeting of unintended sequences in maize although this was not clearly demonstrated in this study. The aflR binding motifs are conserved in the zinc-finger DNA-binding domains in fungi but the transcription factor has also been shown to bind to deviated sequences rather than the typical motifs in fungi (Yu 2012). At the mRNA level, we did not identify any aflR homolog in maize using BLAST searches. The lack of potential off-targets in the two only identified ethylene responsive factors in maize made it difficult to interpret the distorted maize phenotype. In conclusion, our study successfully demonstrated the downregulation of aflatoxin biosynthesis and accumulation in maize. The level of aflatoxins accumulating in transgenics was just at the threshold for the limit permitted in food by KEBS as well as American Food and Drug administration (Van Egmond et al. 2007). Although the gene targeted resulted into other non-intended effects on the host plant, it opens a frontier where other regions of aflR or other genes in the aflatoxin biosynthetic pathway could be targeted using the technology.
Author contribution statement
J. O. M carried out the experiments, participated in vector development, interpretation of results and manuscript preparation. J. M. M participated in vector development, carrying out of experiments and interpretation of results. R. A. O participated in tissue culture and drafting of the manuscript. S. C. O participated in the bioinformatics analyses and drafting of the manuscript. E. O. M participated in coordination, mycotoxin analysis and interpretation of the results. A. E. A conceived the study, designed the experiments, vector development, interpretation of results and manuscript preparation. All authors read and approved the final draft of the manuscript.
References
Alakonya AE, Monda EO (2013) A new approach in aflatoxin management in Africa: targeting aflatoxin/sterigmatocystin biosynthesis in Aspergillus species by RNA silencing technique. Recent Adv Future Prospects. doi:10.5772/51440
Alakonya A, Kumar R, Koenig D, Kimura S, Townsley B, Runo S, Garces HM, Kang J, Yanez A, David-Schwartz R, Machuka J, Sinha N (2012) Interspecific RNA interference of SHOOT MERISTEMLESS-Like disrupts Cuscuta pentagona plant parasitism. Plant Cell 24:3153–3166
Amaike S, Keller NP (2011) Aspergillus flavus. Annu Rev Phytopathol 49:107–133
Atanda OO, Akpan I, Enikuomehin OA (2006) Palm kernel, an alternative culture medium for rapid detection of aflatoxins in agricultural commodities. Afri J Biotech 5:1029–1033
Atanda OO, Ogunrinu MC, Olurunfemi FM (2011) A neutral red desiccated coconut agar for rapid detection of aflatoxigenic fungi and visual determination of aflatoxins. World Mycotoxin J 4(2):147–155
Baulcombe D (2004) RNA silencing in plants. Nature 431:356–363
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16:497–516
Bragulat MR, Abarca ML, Caban˜es FJ (2001) An easy screening method for fungi producing ochratoxin A in pure culture. Int J Food Microbiol 71:139–144
Brown DW, Yu JH, Kar H, Fernandes M, Nesbitt TC, Keller NP, Adams TH, Leonard TJ (1996) Twenty five coagulated transcripts define Sterigmatocystin gene cluster in Aspergillus nidulans. Proc Natl Acad Sci USA 93:1418–1422
Cary JW, Montalbano BG, Ehrlich KC (2000) Promoter elements involved in the expression of the Aspergillus parasiticus aflatoxin biosynthesis pathway gene avnA. Biochim Biophys Acta 1491:7–12
Duan CG, Chun-Han W, Hui-Shan G (2012) Application of RNA silencing to plant disease resistance. Science 3:5
Hamilton AJ, Baulcombe DC (1999) A species of small antisense RNA in post transcriptional gene silencing in plants. Science 286:950–952
Hammond SM, Caudy AA, Hannon GJ (2001) Post-transcriptional gene silencing by double-stranded RNA. Nat Rev Genet 2:110–119
Huvenne H, Smagghe G (2010) Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: a review. J Insect Physiol 56:227–235
Ishida Y, Hiei Y, Komari T (2007) Agrobacterium-mediated transformation of maize. Nat Protoc 2:1614–1621
Jackson AL, Linsley PS (2004) Noise amidst the silence: off-target effects of siRNAs? Trends Genet 20(11):521–524
Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21:635–637
Koch A, Kumar N, Weber L, Keller H, Imani J, Kogel KH (2013) Host-induced gene silencing of cytochrome P450 lanosterol C14ademethylase-encoding genes confers strong resistance to Fusarium species. Proc Natl Acad Sci USA 110:19324–19329
Lewis L, Onsongo M, Njapau H, Schurz-Rogers H, Luber G, Kieszak S, Nyamongo J, Backer L, Dahiye AM, Misore A (2005) Aflatoxin contamination of commercial maize products during an outbreak of acute aflatoxicosis in eastern and central Kenya. Environ Health Perspect 113:1763–1767
Magan N, Abdel-Hadi AM, Caley DP, Carter DRF (2011) Control of aflatoxin production of Aspergillus flavus and Aspergillus parasiticus using RNA silencing technology by targeting aflD (nor-1) gene. Toxins 3:647–659
McDonald T, Brown D, Keller NP, Hammond TM (2005) RNA silencing of mycotoxin production in Aspergillus and Fusarium species. Mol Plant Microbe Interact 18:539–545
Micali CO, Neumann U, Grunewald D, Panstruga R, O’Connell R (2011) Biogenesis of a specialized plant-fungal interface during host cell internalization of Golovinomyces orontii haustoria. Cell Microbiol 13:210–226
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Mutegi CK, Ngugi HK, Hendriks SL, Jones RB (2009) Prevalence and factors associated with aflatoxin contamination of peanuts from Kenya. Int J Food Microbiol 130:27–34
Niu QW, Lin SS, Reyes JL, Chen KC, Wu HW, Yeh SD (2006) Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat Biotech 24:1420–1428
Nowara D, Gay A, Lacomme C, Shaw J, Ridout C, Douchkov D, Hensel G, Kumlehn J, Schweizer P (2010) HIGS: host-induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis. Plant Cell 22:3130–3141
Nunes CC, Dean RA (2012) Host-induced gene silencing: a tool for understanding fungal host interaction and for developing novel disease control strategies. Mol Plant Pathol 13:519–529
Payne GA, Nystrom GJ, Bhatnagar D, Cleveland TE, Woloshuk CP (1993) Cloning of the afl-2 gene involved in aflatoxin biosynthesis from Aspergillus flavus. Appl Environ Microbiol 59:156–162
Pitt JI, Hocking AD (1997) Fungi and food spoilage, 2nd edn. Blackie Academic and Professional, London
Saxena S, Jonsson ZO (2003) Small RNAs with imperfect match to endogenous mRNA repress translation: implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem 278(45):44312–44419
Snove O, Holen T (2004) Many commonly used siRNAs risk off-target activity. Biochem Biophys Res Commun 391(1):256–263
Tinoco ML, Dias BB, Dall’Astta RC, Pamphile JA, Aragão FJ (2010) In vivo trans-specific gene silencing in fungal cells by in planta expression of a double-stranded RNA. BMC Biol 31:27
Van Egmond HP, Schothorst RC, Jonker MA (2007) Regulations relating to mycotoxins in food: perspectives in a global and European context. Anal Bioanal Chem 389:147–157
Voegele RT, Mendgen K (2003) Rust haustoria: nutrient uptake and beyond. New Phytol 159:93–100
Wesley SV, Helliwell CA, Smith NA, Wang MB, Rouse DT, Liu Q, Gooding PS, Singh SP, Abbott D, Stoutjesdijk PA, Robinson SP, Gleave AP, Green AG, Waterhouse PM (2001) Construct design for efficient, effective and high throughput gene silencing in plants. Plant J 27:581–590
Windham GL, Williams WP (2007) A comparison of inoculation techniques for inducing aflatoxin contamination and Aspergillus flavus kernel infection on corn hybrids in the field. Phytoparasitica 35(3):244–252
Xu P, Zhang Y, Kang L, Roossinck MJ, Mysore KS (2006) Computational estimation and experimental verificationof off-target silencing during posttranscriptional gene silencing in plants. Plant Physiol 142:429–440
Yin C, Jurgenson JE, Hulbert SH (2011) Development of a host-induced RNAi system in the wheat stripe rust fungus Puccinia striiformis. Mol Plant Microbe Interact 24:554–561
Yu J (2012) Current understanding on aflatoxin biosynthesis and future perspective in reducing aflatoxin contamination. Toxins 4:1024–1057
Yu J, Bhatnagar D, Cleveland TE (2004) Completed sequence of aflatoxin pathway gene cluster in Aspergillus parasiticus. FEBS Lett 564:126–130
Zidani S, Ferchichi A, Chaieb M (2005) Genomic DNA extraction method from pearl millet (Pennisetum glaucum) leaves. Afr J Biotech 4(8):862–866
Acknowledgments
The authors wish to acknowledge financial support from the Bill and Melinda Gates Foundation through the Grand Challenge initiative (Grant No: OPP1058537) to Dr. Alakonya.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Benvenuto.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Masanga, J.O., Matheka, J.M., Omer, R.A. et al. Downregulation of transcription factor aflR in Aspergillus flavus confers reduction to aflatoxin accumulation in transgenic maize with alteration of host plant architecture. Plant Cell Rep 34, 1379–1387 (2015). https://doi.org/10.1007/s00299-015-1794-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-015-1794-9