
Abstract
Key message
MicroRNAs have higher expression stability than protein-coding genes in B. napus seeds and are therefore good reference genes for miRNA and mRNA RT-qPCR analysis.
Abstract
Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) has become the “gold standard” to gain insight into function of genes. However, the accuracy of the technique depends on appropriate reference genes for quantification analysis in different experimental conditions. Accumulation of microRNAs (miRNAs) has also been studied by RT-qPCR, but there are no reference genes currently validated for normalization of Brassica napus miRNA expression data. In this study, we selected 43 B. napus miRNAs and 18 previously validated mRNA reference genes. The expression stability of the candidate reference genes was evaluated in different tissue samples (stages of seed development, flowers, and leaves) using geNorm, NormFinder, and RefFinder analysis. The best-ranked reference genes for expression studies during seed development (miR167-1_2, miR11-1, miR159-1 and miR168-1) were used to asses the expression of miR03-1. Since candidate miRNAs showed higher expression stability than protein-coding genes in most of the tested conditions, the expression profile of DGAT1 gene was compared when normalized by the four most stable miRNAs reference genes and by the four most stable mRNA reference genes. The expected expression pattern of DGAT1 during seed development was achieved with the use of miRNA as reference genes. In conclusion, the most stable miRNA reference genes can be employed in the normalization of RT-qPCR quantification of miRNAs and protein-coding genes. This work is the first to perform a comprehensive survey of the stability of miRNA reference genes in B. napus and provides guidelines to obtain more accurate RT-qPCR results in B. napus seeds studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brassica napus, known as rapeseed, is one of the most important crops for the production of vegetable oil, biofuel, and animal feed in the world. The demand for vegetable oil is increasing in recent years, and therefore seeds with improved oil content and quality are the major challenges for genetic improvement of oilseed crops (Lu et al. 2011). Modification of the fatty acid composition of oilseed crops has been achieved through genetic manipulation of enzyme-catalyzed reactions in fatty acid and in triacylglycerol biosynthesis. There has also been considerable interest in modifying the action of transcription factors that can up-regulate a series of biochemical reactions in carbon flow (Weselake et al. 2009). Further understanding of the regulatory mechanisms of seed development is essential to identify the molecular basis of lipid deposition during seed development.
The small non-coding RNAs microRNAs (miRNAs) regulate transcriptionally and post-transcriptionally the expression of several genes and are emerging as important regulators of seed development (Nodine and Bartel 2010; Willmann et al. 2011). In fact, the majority of known plant miRNAs appear to target transcription factor genes, which position miRNAs in the key role of establishing organ patterns and fertility in developing plants. Although the role of miRNA-regulated gene expression in plant biology is just beginning to be appreciated, they could be a useful tool in breeding programs for seed quality traits. The advances in genome and transcriptome sequencing have contributed to the identification of a wide range of small noncoding RNAs, especially miRNAs, from different plant species. Several new miRNAs and its target genes were recently identified in seeds of B. napus using deep sequencing (Huang et al. 2013; Körbes et al. 2012; Zhao et al. 2012). However, functional studies during seed development will require miRNA quantitation for a myriad of downstream applications, including validation of predicted miRNAs, tissue expression profiling, and comparison with mRNA expression or biomarker discovery. Changes in miRNA levels can be modest, and yet have profound cellular consequences. As a result, precise measurements of miRNA expression are critically important.
The short sequences and high similarity within large miRNA gene families make miRNAs difficult and unreliable targets for traditional hybridization-based detection methods. On the contrary, RT-qPCR is a powerful tool to accurately and quickly validate the expression levels of miRNAs and its target genes. This technique, based on relative quantification method, normalize the expression level of a gene of interest to the expression level of a control gene, called reference genes (Bustin et al. 2009; Derveaux et al. 2010; Rieu and Powers 2009). Therefore, it is of paramount importance the selection of robust control genes for reliable results (Guénin et al. 2009; Livak and Schmittgen 2001). An ideal reference gene is expected to present a constant expression level through different conditions, such as plant developmental stages and tissue types and its expression should be unaffected by experimental parameters (Vandesompele et al. 2002). Studies suggested that several reference genes should be used as internal standards because the use of a single gene for normalization could lead to large errors (Udvardi et al. 2008). To select appropriate candidate reference genes, it is highly recommended to test several control genes, such as endogenous miRNA genes and housekeeping genes, so that at least two can be chosen to normalize the miRNA experiment.
The most commonly housekeeping genes used to normalize RT-qPCR data are cytoskeleton components (actins) and genes involved in basic cellular processes, such as glycolysis [(glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], protein folding (cyclophilin), synthesis of ribosomal subunits (rRNA), electron transport, and protein degradation (ubiquitin). Many of these reference genes have been validated for vegetative tissues, but recent findings demonstrated that they were not stably expressed in seeds and pollen (Paolacci et al. 2009; Wei et al. 2010). Other studies have shown the same inconsistencies with the most common references genes under different experimental conditions (Czechowski et al. 2005; Dekkers et al. 2012; Gutierrez et al. 2008b). Plant endogenous miRNAs have been established for soybean, wheat, longan tree, citrus and lettuce (Kulcheski et al. 2010; Feng et al. 2012; Kou et al. 2012; Lin and Lai 2013; Borowski et al. 2014). However, it has been shown that a good reference gene from one species does not necessarily have well stability in another species (Gutierrez et al. 2008a). To date few studies evaluated reference genes in B. napus and were mainly focused on protein-coding genes expression in vegetative tissues (Chen et al. 2010; Wang et al. 2014; Yang et al. 2014). Therefore, it is necessary to determine appropriate reference genes for the study of miRNAs in B. napus seeds.
In the present study, we have compared the expression stability of 43 miRNAs and 18 protein-coding mRNAs commonly used for normalization in different stages of seed development, in flower and leaf samples. Our results revealed a group of four miRNAs that should be used for accurate normalization of miRNA expression in seeds. Further, the expression stability of miRNAs was higher than protein-coding genes and therefore miRNAs can also be used as optimal reference genes in RT-qPCR analysis of protein-coding genes.
Materials and methods
Plant material
Brassica napus (cultivar PFB-2, Embrapa) plants were grown in open field (30°S 51 W) from April to September 2011 in Porto Alegre, Brasil. Flowers, young leaves, and seeds were collected in quadruplicates. Each sample was composed by a pool of tissues from five different plants. Seeds dissected from siliques were collected at 7, 14, 21, 28 and 35 days after flowering (DAF). All samples were immediately frozen in liquid nitrogen and stored at −80 °C until RNA isolation.
Total RNA isolation
Total RNA was extracted from each biological replicate using the NucleoSpin® RNA Plant kit, following the manufacturer's recommendation (Macherey–Nagel, Germany). Residual genomic DNA contamination was removed by on-column digestion with RNase-free DNase set supplied with the extraction kit. RNA integrity and the absence of genomic DNA was evaluated by electrophoresis on 1.2 % agarose gel. RNA quantification and OD 260/280 ratios were determined using a Nanodrop Spectrophotometer (Thermo Scientific, Wilmington, USA).
Selection of candidate reference genes and primer design
In a previous study of the miRNA transcriptome of mature and developing B. napus, we identified sets of miRNAs which were equally or differentially represented in the two small RNA-seq libraries (Körbes et al. 2012). From this study, we chose a set of 43 miRNAs which were abundantly present on all seed samples. Specific stem–loop primers were designed for miRNA cDNA synthesis, as previously described (Chen et al. 2005). The stem-loop sequence consists of 44 conserved and 6 variable nucleotides (N) that are specific to the 3′ end of each miRNA sequence (5′GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACNNNNNN 3′). Specific forward primers for each full miRNA sequence were designed (Supplementary Table 1) and used in combination with a universal reverse primer (5′GTGCAGGGTCCGAGGT 3′) for RT-qPCR amplifications (Chen et al. 2005).
For protein-coding genes, a set of 18 protein-coding genes were evaluated as candidate reference genes. Eight protein-coding genes, (PP2A, TIP41, SAND, UP2, UBC21, ACT7, UBC9 and UP1), were selected from a previous report of reference genes for RT-qPCR analysis of vegetative tissues and developing embryos of B. napus (Chen et al. 2010). In addition, ten protein-coding genes (ASAR, ACT2 TUA5, UKN1, UKN2, TPR1, MPT, HDC, PEPKR1, GAPDH) were selected based on previous works with peanuts and tomato seeds (Chi et al. 2012; Dekkers et al. 2012). The reported sequences were used as query for Blastx in NCBI Database (http://www.ncbi.nlm.nih.gov) and the retrieved putative orthologs in B. napus are listed on Supplementary Table 2. Primer pairs were designed with B. napus sequences, using the online tool Primer3 (http://frodo.wi.mit.edu/primer3/) with the following parameters: Tm around 60 °C and product size range 120–150 base pairs, primer sequences with a length of 19–21 nucleotides with an optimum at 20 nucleotides, and a GC content of 45–55 %. All primer sequences are listed in Supplementary Table 1.
cDNA synthesis and RT-qPCR conditions
RNA was reverse transcribed using multiplex technique from approximately 1 µg of total RNA (Lao et al. 2006; Tang et al. 2006). Each cDNA synthesis reaction was primed with a pool of 2.3 µM 43 miRNA-specific stem–loop primers. For mRNA cDNA synthesis, 1 µM oligonucleotide dT24V was used. Before transcription, RNA and primers were mixed with RNase-free water, to a total volume of 10 µL, and incubated at 70 °C for 5 min, followed by ice cooling. Then, 6 µL 5× RT-Buffer (Promega), 3 µL of 5 mM dNTP (Ludwig Biotecnologia LTDA), and 1.5 µL MML-V RT enzyme 200 U (Promega) were added for a final volume of 30 µL. The synthesis reaction was performed at 16 °C for 30 min on a Veriti Thermal Cycler (Applied Biosystem), 37 °C for 30 min and inactivation of the enzyme was completed at 85 °C for 5 min. All cDNA samples were 100-fold diluted with RNase-free water before used as a template in RT-qPCR analysis.
RT-qPCR reactions were performed in a CFX384 Real-Time PCR Detection System (Bio-Rad). Reaction’s volume of 10 µL contained 5 µL of diluted cDNA (1:100), 0.1× SYBR Green I (Invitrogen), 2.5 µM dNTP, 1× PCR Buffer, 3 mM MgCl2, 0.25 U Platinum Taq DNA Polymerase (Invitrogen), and 0.2 µM of each reverse and forward primers. Samples were analyzed as technical quadruplicates and no template controls were included. The PCR cycling conditions were: 95 °C for 5 min, 40 cycles of 94 °C for 15 s, 60 °C for 10 s, then 72 °C for 10 s. Melting curves were performed immediately after the completion of the RT-qPCR and fluorescence measured from 65 to 99 °C. Threshold was manually determined using the CFX384 Manager Software (Bio-Rad).
Data analysis
Descriptive statistics were performed in excel. Primer efficiency (E) and correlation coefficient (R 2) were calculated from amplification curves using LinRegPCR (Ramakers et al. 2003; Ruijter et al. 2009). The software determines the baseline fluorescence and does a baseline subtraction. Then a Window-of-Linearity is set and PCR efficiencies per sample are calculated. The statistical software geNorm was used to determine the most stable reference genes and to determine the optimal number of genes required for normalization (Vandesompele et al. 2002). geNorm determines a measure of gene expression stability (M value) first and, via a stepwise exclusion of the less stable gene, creates a stability ranking. A valuable feature of geNorm output is the V value that reflects the minimal number of genes required for a robust normalization factor (NF). It starts calculating the pairwise variation (V) V2/3 between the NF2 (including the two most stable reference genes) and the normalization factor NF3 (including the three most stable reference genes). Then, it performs a stepwise calculation of the Vn/n + 1 between the NFn and the NFn +1. Gene expression stability of candidate reference genes was also analysed by NormFinder and three additional statistical approaches implemented in RefFinder (BestKeeper, Delta CT and Comprehensive Ranking) (http://www.leonxie.com/referencegene.php). NormFinder uses an ANOVA-based model to estimate intra- and inter-group variation and combines these estimates to provide a direct measure of the variation in expression for each gene (Andersen et al. 2004). BestKeeper determines the stability of the gene candidates based on the SD of the gene expression levels across samples. Then, genes with least variable cycle quantification (Cq) values are used for pairwise correlation analysis to an optimal normalization factor that merges data from all of them. The geometric mean of the Cq values of the most highly correlated genes is used to calculate a BestKeeper index, which is followed by Pearson’s correlation coefficient to determine the similarity in the expression levels among the candidates. Thus, genes with least SDs and highest correlation with the index are ranked as the most stable genes (Pfaffl et al. 2004). Delta CT (dCt) method depends on a concept similar to that of geNorm where genes within one sample are compared to each other by calculating the dCq value. The gene pair with the same dCt value (smallest SD) across all samples is considered to be stable and vice versa. The average of all SD values for each gene set of pairwise combinations is used to rank the gene stability. Genes with lower average SD are more stable than others. RefFinder is a web-based interface that calculates a comprehensive rank based on the most stable gene candidates among all methods. For each gene, RefFinder calculates the geometric mean of the ranks calculated by each of the previous approaches. Genes with the lowest rank geometric mean are considered as most stable.
Reference gene validation
The expressions of two genes, one miRNA (miR03-1) and one protein-coding gene were normalized with the most or the following three most stable miRNA reference genes selected by geNorm analysis. The protein-coding gene diacylglycerol O-acyltransferase 1 (DGAT1) (Forward 5′ GTATACTTTCCGTGCCTGCGCATC 3′; Reverse 5′ CACCATGGAGCCAAACCTTTCTTGTA 3′) was selected due to its importance for lipid storage during seed development. DGAT1 was also normalized with the most or the following four most stable mRNA genes selected by geNorm. To calculate the relative expression of the two target genes in triplicate samples, we used the 2−∆∆Ct method (Livak and Schmittgen 2001). Samples at 7 DAF were used as the reference samples to calculate the relative expression levels of miR03-1 and DGAT1 target genes during different stages of seed development.
Results
PCR efficiency and amplification specificity
A comprehensive set of 61 candidate genes, including 43 miRNAs and 18 protein-coding genes, were selected based on previous reports (Körbes et al. 2012; Chen et al. 2010; Chi et al. 2012; Dekkers et al. 2012). First the amplification efficiency for each specific primer-pair was determined, since optimal amplification conditions are absolutely essential for accurate and reproducible quantification of samples. The hallmarks of an optimized RT-qPCR assay are: linear standard curve with coefficient of correlation R 2 > 0.980 and amplification efficiency (E) that ranges from 1.8 to 2.1. Ideally, the efficiency of a PCR should be 100 % (E = 2), meaning that for each cycle the amount of product doubles. According to the criteria, the amplification of nine candidate genes, miR167-2_6, miR158-16, miR164_51, miR393-1_2, miR172-1, miR319-130, miR09-1, miR396-164 and miR400-185 did not meet the minimum requirements and were excluded from further analysis. Three additional candidate genes (miR319-132, miR05_1 and miR408-1) were excluded due to inconsistent amplification within biological quadruplicates. The amplification efficiency values of the remaining protein-coding and miRNAs candidate reference genes ranged within acceptable limits, as listed in Supplementary Table 1. The results indicate that 49 primer pairs are suitable for PCR reactions of high quality and specificity and therefore were further evaluated.
Expression stability analysis and ranking of candidate reference genes
Gene expression of the 49 candidate genes was analyzed on four different datasets: (1) all tissues, including flower, leaf, and five stages of seed development; (2) five stages of seed development; (3) flower samples; and (4) leaf samples. The RT-qPCR data were analyzed by geNorm in order to evaluate the expression stability and to determine the minimal number of genes required for normalization for each dataset. Four additional statistical approaches (NormFinder, BestKeeper, Delta CT and RefFinder) were used to evaluate candidate reference gene stability as an effort to minimize intrinsic bias relative to each approach.
geNorm analysis
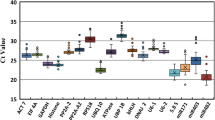
geNorm is the most common algorithm used to assess gene expression stability and it ranks the reference genes by calculating the gene expression stability value M, based on the average pairwise expression ratio. The lowest M value indicates the most stable expression, whereas the highest M value indicates the least stable expression. The program recommends using M value below the threshold of 1.5 to identify reference genes with stable expression and therefore candidate genes with M > 1.5 were excluded from dataset analysis. Cycle quantification (Cq) values of each candidate gene that was further evaluated were analyzed to describe the steady-state mRNA levels in 24 samples of three different tissues. The candidate reference genes showed a relative wide range of Cq values and individual genes presented different expression levels through all samples tested (Fig. 1). The lowest mean Cq value was observed for miR390-144 (12.24) in seeds (Fig. 1b), and the highest Cq value was exhibited by TPR1 (29.39) in all tissues (Fig. 1a). In general, we could observe that miRNAs presented lower Cq values and smaller variation than protein-coding genes in all analyzed tissues.

Transcript abundance shown by quantitative cycle values (Cq) of candidate genes analysed by geNorm in four datasets: a all tissues, b five stages of seed development, c flowers, d leaves. The solid line represents the median value, and the boxes are 25th and 75th percentiles. Whisker caps represent the maximum and minimum Cq values
geNorm analysis showed that average M values of miRNAs were lower than the M values of protein-coding genes (Fig. 2). The gene pair miR159-1 and miR168-1 were the most stable combination of genes in all tissues dataset, with M value of 0.49 (Fig. 2a). For the seed dataset, miR167-1_2 and miR11-1 were the most stable combination of all genes tested (M = 0.52) (Fig. 2b). For flower samples, miR156-1_1 and miR168-1 are the most stable combination for reference genes (M = 0.18) (Fig. 2c). An exception was observed in the leaf dataset, where protein-coding genes UBC9 and UBC21 were the most stable combination recommended for gene normalization (M = 0.15) (Fig. 2d). Nevertheless, miRNAs showed M values below 0.8, which indicates that they also show high expression stability in leaves.

Gene expression stability and determination of the optimal number of reference genes by geNorm. Average expression stability values (M) in a all tissues dataset, b five different stages of seed development, c flower and d leaf samples. e Optimal number of reference genes required for effective normalization in each dataset
For accurate analysis of RT-qPCR results, it is highly recommended the use of at least two reference genes (Vandesompele et al. 2002). To evaluate the optimal number of genes required for accurate normalization, we used geNorm tool to calculate the pairwise variation (V) for all the four datasets. Values below the V cutoff (0.15) indicate the optimal number of control genes for reliable normalization. The observed pairwise variation V2/3 was 0.165 and 0.169 for all tissues and for developing seeds, respectively (Fig. 2e). Strictly applying the cutoff of 0.15 in all tissues dataset (V3/4 = 0.145) and seed dataset (V3/4 = 0.135), indicate that at least three reference genes should be included for gene expression studies in both groups of data. In flower and leaf datasets, the combination of the two most stable genes would be sufficient for normalization purposes, because the V2/3 value was lower than 0.15 (Fig. 2e). Additionally, using a combination of the four, five and six most stable genes, the V value still remains below the cutoff value (0.15). Increasing the number of reference genes until nine (V8/9) could contribute to the accuracy of normalization of data in all tissues and seed samples, but the use of ten or more reference genes cannot contribute to the improvement of normalization accuracy. geNorm analyses indicate the combination of miR159-1, miR168-1, and miR156-3_3 for accurate normalization of all tissues dataset. For gene expression studies comparing stages of seed development, the combinations of miR11-1, miR167-1_2, and miR159-1 are recommended, whereas the miR156-1_1 and miR168-1 are suitable for flower dataset. The mRNA UBC9 and UBC21 are recommended for leaf RT-qPCR data normalization (Fig. 2e).
NormFinder analysis
NormFinder is another commonly used algorithm. It is based on variance estimation and identifies the optimal normalization genes among a set of candidate genes under a given set of experimental conditions. In contrast to geNorm, NormFinder considers intra- and inter-group variations when calculating the normalization factor (NF). Candidate genes with minimal intra- and inter-group variation will have the lowest stability value and will be ranked at the top. We used NormFinder to analyze gene expression stability of 49 candidate reference genes and the results are summarized in Supplementary Table 3 and Supplementary Fig. 1. The ranking generated by this approach for all tissues and developing seed datasets was somewhat different from the one determined by geNorm. Despite being an altered rank order, the same ten genes indicated for all tissues dataset were the most stable genes identified by both NormFinder and geNorm programs. A similar result was found for the seed dataset with the top 20 genes from both NormFinder and geNorm programs. In fact, the 10 best-ranked genes showed small differences in their stability values within all tissues set and seed set (Supplementary Table 3; Supplementary Fig. 1a). As observed in geNorm analyses, miRNAs showed generally smaller stability values and were ranked at the top in all sample datasets (Supplementary Fig. 1a). When compared to geNorm, a visible variation was observed in the order of NormFinder rankings of flower and leaf datasets (Supplementary Fig. 1c, d).
RefFinder analysis
Since the gene ranks of geNorm and NormFinder were not exactly the same, we used RefFinder analyses to obtain additional ranks of the best candidate genes with other available methods. RefFinder takes into consideration the rankings of geNorm, NormFinder, BestKeeper, and Delta Ct methods to recommend a comprehensive ranking of the most stable genes. BestKeeper results were based on the descriptive statistics described in Supplementary Table 4. The top 10 candidate genes for all tissues and seed datasets from each approach are presented in Table 1. The results for flower and leaf datasets are shown in Supplementary Table 5. Briefly, RefFinder analyses ranked miR156-3_3, miR159-1, miR168-1, miR156-1_1, and miR11-1 as the five best reference genes for all tissues samples. In the dataset of developing seeds, miR156-3_3, miR159-1, miR156-7, miR11-1, and miR167-1_2 were identified as the top five genes. The top three genes ranked by geNorm in all tissues and developing seeds were also present in the top three and top five genes ranked by RefFinder, respectively (Table 1). Despite the different ordinal rank, the analyses indicate that the geNorm top four genes are reliable reference genes for RT-qPCR normalization in seed samples.
Validation of putative reference genes
The use of inaccurate reference genes to calculate relative expression data could have a significant influence on the final normalized results. geNorm analysis returns the best combination and the number of genes required for reliable normalization. To validate the miRNA reference gene set recommended by geNorm, we evaluate the expression of the B. napus specific miR03-1 in five seed development stages (Fig. 3). miR03-1 was selected as one of the putative stably expressed genes in small RNA-seq experiments and has an unknown expression profile during the development of B.napus seeds. In addition, we tested the impact of increasing the number of reference genes in the normalization of miR03-1. Small differences in the expression of this miRNA were expected, since it was one of the miRNAs selected as candidate reference gene (Supplementary Table 1). The use of a single gene lead to a wide variation in the relative expression of miR03-1 and to larger variation of the standard error, which was reduced by the addition of one or more reference genes.

Relative expression of miR03-1 in five different stages of seed development after normalization with geNorm reference genes miR167-1_2/miR11-1/miR159-1/miR168-1. Seed samples at 7 DAF were used as reference samples. The error bar shows the mean standard error calculated from three biological replicates
We observed that miRNAs have better expression stability than protein-coding genes in seed samples. To evaluate the use of miRNA reference gene set for normalization of an mRNA, we monitored by RT-qPCR the expression patterns of diacylglycerol O-acyltransferase 1 (DGAT1) during seed development (Fig. 4). DGAT1 is an enzyme involved in the last step of triacylglycerol biosynthesis in seeds. The expression pattern of DGAT1 in developing oilseeds usually increases during seed filling stages followed by a decrease when the seed reaches maturation (Troncoso-Ponce et al. 2011; Li et al. 2013). When DGAT1 was normalized with the combination of miRNA genes, the expression pattern was consistent to the relative expression patterns reported in B. napus (Fig. 4a). A different result was observed when DGAT1 was normalized using the best combination of protein-coding candidate reference genes of geNorm (Table 1). In this case, the expression of DGAT1 was higher in the third and fifth development stages when compared to miRNAs normalized data (Fig. 4a). The impact on DGAT1 normalization with one, two, three, or four most stable miRNA or protein-coding reference genes was also evaluated (Fig. 4b, c). In both cases, the expression patterns were not altered, but high standard error values were observed when less reference genes were used to normalize expression. This result shows that at least three reference genes are necessary to produce accurate detection of expression levels among different seed samples. Most interestingly, the results indicate that miRNAs are also suitable reference genes in accurate normalization of protein-coding genes.

Relative expression of DGAT1 in five different stages of seed development after normalization with geNorm reference genes: a miR167-1_2/miR11-1/miR159-1/miR168-1 and TIP41/UBC21/GAPDH/UP2, b individual and/or combined miRNA reference genes, c individual and/or combined protein-coding (mRNAs) reference genes. Seed samples at 7 DAF were used as reference samples. The error bar shows the mean standard error calculated from three biological replicates
Discussion
Quantitative real-time PCR is a powerful technique for protein-coding gene and miRNA expression studies and relies on the critical procedure of data normalization for accurate and reliable output (Bustin et al. 2009). RT-qPCR studies in seeds are challenging, because during development seeds go through a complex network of many dynamic developmental, biochemical, and metabolic processes (Baud and Lepiniec 2010). During this phase, the expression of several hundreds of genes involved in spatial and temporal regulation of these metabolic processes is globally affected (Le et al. 2010). Due to these characteristics, it is difficult to select optimal reference genes for RT-qPCR assays in seed tissues. To date few studies have focused on selecting suitable reference genes for seed tissues in crop plants and none considered miRNAs quantification (Chen et al. 2010; Dekkers et al. 2012; Han et al. 2012; Li et al. 2012). With this in mind, the present work performed comprehensive analysis candidate reference genes for miRNA gene expression studies B. napus seeds.
A total of 61 miRNA and mRNA candidate genes were selected to perform comprehensive expression stability analysis in different tissues. The performance of each candidate gene was tested in different samples (developing seeds, flower and leaf tissues) to help define suitable sets of reference genes for expression profiles studies in seeds, but also to allow comparative studies of seeds with other reproductive or vegetative tissue. The commonly used geNorm and NormFinder algorithms were used to identify the best suitable reference gene sets for each group of tissue sample. Although the results from the two algorithms were largely similar, they did not rank genes in the exactly same order. These ranking discrepancies were previously reported (Kulcheski et al. 2010) and reflect differences in the calculation method of these two approaches. In addition, a drawback of NormFinder is the requirement of a minimum of 8 samples/group. Therefore, the observed discrepancies in the flower and leaf datasets could be attributed to differences in program methodologies and the small sample size of flower and leaf dataset (n = 4). When we used the web-based tool RefFinder to access expression stability with additional methods we observed that miRNAs were also among the top 10 most stably expressed candidates in all different methods and sample groups, except in leaf dataset (Supplementary Fig. 5). A small difference in NormFinder rank for all tissue sample set was observed when it was calculated by RefFinder, but the most stable genes identified in Supplementary Table 3 where the same top 10 genes identified by RefFinder (Table 1). This difference could be caused by the algorithm used by RefFinder but clearly did not compromise the identification of the most stable genes.
We concluded that geNorm results are thus reliable and therefore we recommend the use of miR167-1_2, miR11-1, and miR159-1 as the three most suitable reference genes for seed samples and the use of miR167-1_2, miR11-1, and miR159-1 when comparing gene expression in seeds, flower, and leaves. Although miRNAs were not well ranked in leaf dataset by any method, geNorm and NormFinder show that miRNA stability values were similar to protein-coding genes. Therefore, we conclude that miRNAs are probably good reference genes also for leaf samples (Fig. 1; Supplementary Fig. 1). However, we strongly advise the prospective user to perform further RT-qPCR validation in experimental setups where only leaf tissues are used before confidently using miRNAs as reference genes. Nevertheless, we have validated the stable expression of a sufficient number of miRNAs for experimental setup with seeds only or combined with flowers and leaves.
The miRNA candidate reference genes selected for the current study performed better than the previous validated protein-coding genes. Recent studies in other plant species have also demonstrated that miRNAs showed higher expression stability than protein-coding genes under stress treatments (Feng et al. 2012; Kulcheski et al. 2010; Borowski et al. 2014) and during somatic embryogenesis (Lin and Lai 2013). In addition, Kulcheski et al. (2010) demonstrated that miRNAs are optimal reference genes not only for other miRNAs but also for protein-coding genes in RT-qPCR analysis. Similar results were also obtained in studies searching for suitable reference genes for the normalization of miRNA expression on humans and porcines (Peltier and Latham 2008; Timoneda et al. 2012). Therefore, the group of miRNA reference genes indicated by geNorm analysis was validated not only by performing normalization of the expression of a new miRNA specific to B. napus but also with a protein-coding gene.
DGAT1 is an important gene for lipid biosynthesis and its expression profile has been described in developing seeds of oilseed species (Troncoso-Ponce et al. 2011; Li et al. 2013). geNorm analysis ranked TIP41, UBC21, GAPDH, and UP2 genes as the best protein-coding reference genes for developing seeds dataset. In previous reports, UBC21 and TIP 41 were the most stable genes among different stages of embryo development in B. napus (Chen et al. 2010), and UBC protein was reported as a stable reference gene during developing embryos in different cultivars of Tung (Han et al. 2012). It is known that the use of less stable reference genes, in this case protein-coding genes, can generate biases that could lead to misinterpretation of gene expression patterns. Using different normalization strategies of DGAT1 gene (with miRNAs or mRNAs), we show that miRNAs produced similar results to protein-coding genes. However, the high increase in DGAT1 expression in 21 DAF was not expected when normalized to mRNA (Fig. 4a). The normalization with miRNAs showed a gradual increase in the expression and therefore we considered them as appropriate endogenous reference genes for normalization of protein-coding genes in developing seed tissues.
Conclusions
In this work, we focused on establishing reliable reference genes for gene miRNA and its target genes expression studies in developing seed samples. We performed comprehensive analysis with several candidate reference genes in developing seeds, flower, and leaf samples. In conclusion, we recommend the use of at least three reference genes (miR167-1_2/miR11-1/miR159-1) for SYBR Green RT-qPCR assays to compare the expression of miRNAs in seed samples. In addition, we provide evidence that support the use of miRNA reference genes to calculate the expression pattern of protein-coding genes. This discovery can be particularly useful in studies which aim to validate target genes of miRNAs. Furthermore, our study will also benefit future gene RT-qPCR expression studies in other species of the genus Brassica.
Author contribution statement
APK, MMP and RM conceived and designed research. RDM, APC and GLM conducted experiments. RDM, APC, APK and RM analyzed data. RDM and APK wrote the manuscript. All authors read and approved the manuscript.
References
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Baud S, Lepiniec L (2010) Physiological and developmental regulation of seed oil production. Prog Lipid Res 49:235–249
Borowski JM, Galli V, da Silva Messias R et al (2014) Selection of candidate reference genes for real-time PCR studies in lettuce under abiotic stresses. Planta 239:1187–1200
Bustin SA, Benes V, Garson JA et al (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Chen C, Ridzon DA, Broomer AJ et al (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179
Chen X, Truksa M, Shah S, Weselake RJ (2010) A survey of quantitative real-time polymerase chain reaction internal reference genes for expression studies in Brassica napus. Anal Biochem 405:138–140
Chi X, Hu R, Yang Q et al (2012) Validation of reference genes for gene expression studies in peanut by quantitative real-time RT-PCR. Mol Gen Genom 287:167–176
Czechowski T, Stitt M, Altmann T (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
Dekkers BJ, Willems L, Bassel GW et al (2012) Identification of reference genes for RT-qPCR expression analysis in Arabidopsis and tomato seeds. Plant Cell Physiol 53:28–37
Derveaux S, Vandesompele J, Hellemans J (2010) How to do successful gene expression analysis using real-time PCR. Methods 50:227–230
Feng H, Huang X, Zhang Q et al (2012) Selection of suitable inner reference genes for relative quantification expression of microRNA in wheat. Plant Physiol Biochem 51:116–122
Guénin S, Mauriat M, Pelloux J et al (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60:487–493
Gutierrez L, Mauriat M, Guénin S et al (2008a) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol J 6:609–618
Gutierrez L, Mauriat M, Pelloux J et al (2008b) Towards a systematic validation of references in real-time RT-PCR. Plant cell 20:1734–1735
Han X, Lu M, Chen Y, Zhan Z, Cui Q, Wang Y (2012) Selection of reliable reference genes for gene expression studies using real-time PCR in Tung tree during seed development. PLoS One 8:e43084
Huang D, Koh C, Feurtado JA et al (2013) MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genom 14:140
Körbes AP, Machado RD, Guzman F et al (2012) Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS One 7:e50663
Kou SJ, Wu XM, Liu Z, Liu YL, Xu Q, Guo WW (2012) Selection and validation of suitable reference genes for miRNA expression normalization by quantitative RT-PCR in citrus somatic embryogenic and adult tissues. Plant Cell Rep 31(12):2151–2163
Kulcheski FR, Marcelino-Guimaraes FC, Nepomuceno AL et al (2010) The use of microRNAs as reference genes for quantitative polymerase chain reaction in soybean. Anal Biochem 406:185–192
Lao K, Xu NL, Yeung V et al (2006) Multiplexing RT-PCR for the detection of multiple miRNA species in small samples. Biochem Biophys Res Commun 343:85–89
Le BH, Cheng C, Bui AQ et al (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA 107:8063–8070
Li Q, Fan CM, Zhang XM, Fu YF (2012) Validation of reference genes for real-time quantitative PCR normalization in soybean developmental and germinating seeds. Plant Cell Rep 31(10):1789–1798
Li R, Hatanaka T, Yu K, Wu Y, Fukushige H, Hildebrand D (2013) Soybean oil biosynthesis: role of diacylglycerol acyltransferases. Funct Integr Genomics 13(1):99–113
Lin YL, Lai ZX (2013) Evaluation of suitable reference genes for normalization of microRNA expression by real-time reverse transcription PCR analysis during longan somatic embryogenesis. Plant Physiol Biochem 66:20–25
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 25:402–408
Lu C, Napier JA, Clemente TE, Cahoon EB (2011) New frontiers in oilseed biotechnology: meeting the global demand for vegetable oils for food, feed, biofuel, and industrial applications. Curr Opin in Biotechnol 22:252–259
Nodine MD, Bartel DP (2010) MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev 24:2678–2692
Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol 10:11
Peltier HJ, Latham GJ (2008) Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 14:844–852
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Ramakers C, Ruijter JM, Deprez RHL, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Rieu I, Powers SJ (2009) Real-time quantitative RT-PCR: design, calculations, and statistics. Plant Cell 21:1031–1033
Ruijter JM, Ramakers C, Hoogaars WMH et al (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45
Tang F, Hajkova P, Barton SC et al (2006) MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res 34:e9
Timoneda O, Balcells I, Córdoba S et al (2012) Determination of reference microRNAs for relative quantification in porcine tissues. PLoS One 7:e44413
Troncoso-Ponce MA, Kilaru A, Cao X et al (2011) Comparative deep transcriptional profiling of four developing oilseeds. Plant J 68:1014–1027
Udvardi MK, Czechowski T, Scheible WR (2008) Eleven golden rules of quantitative RT-PCR. Plant Cell 20(7):1736–1737
Vandesompele J, De Preter K, Pattyn F et al (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:1–11
Wang Z, Chen Y, Fang H, Shi H, Chen K, Zhang Z, Tan X (2014) Selection of reference genes for quantitative reverse transcription polymerase chain reaction normalization in Brassica napus under various stress conditions. Mol Genet Genomics 289(5):1023–1035
Wei LQ, Xu WY, Deng ZY et al (2010) Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genom 11:338
Weselake RJ, Taylor DC, Rahman MH, Shah S, Laroche A, McVetty PB, Harwood JL (2009) Increasing the flow of carbon into seed oil. Biotech Adv 27:866–878
Willmann MR, Mehalick AJ, Packer RL, Jenik PD (2011) MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant Physiol 155:1871–1884
Yang H, Liu J, Huang S, Guo T, Deng L, Hua W (2014) Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 538(1):113–122
Zhao Y-T, Wang M, Fu S-X et al (2012) Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production- and development-correlated expression and new small RNA classes. Plant Physiol 158:813–823
Acknowledgments
This work was financially supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-CNPq, Brazilian Ministry of Education), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Genoprot-CNPq-MCT No. 559636/2009-1; CNPq-Universal No. 472575/2011-2), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (Agroestruturante-FAPERGS). APK, APC, and GLM were sponsored by research and Ph.D. grants from CAPES. RM and MM were sponsored by research grants from CNPq.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Menossi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
299_2015_1773_MOESM1_ESM.tif
Supplementary Fig. 1 Ranking of candidate reference genes based on NormFinder analysis for (a) stages of seed development, (b) flowers, (c) and leaf samples. Lower stability values indicate more stable gene expression (TIFF 672 kb)
299_2015_1773_MOESM2_ESM.pdf
Supplementary Table 1 Primer sequences and amplicon characteristics of 61 candidate reference genes tested for gene expression normalization (PDF 58 kb)
299_2015_1773_MOESM4_ESM.xlsx
Supplementary Table 3 Ranking of candidate reference genes for all tissues sample set was based on gene expression stability calculated by NormFinder. Each tissue sample set was considered as a group for NormaFinder analysis (XLSX 16 kb)
299_2015_1773_MOESM6_ESM.docx
Supplementary Table 5 Gene expression stability of the best candidate reference genes for flower data set (top) and leaf data set (bottom) as assessed by RefFinder (DOCX 16 kb)
Rights and permissions
About this article

Cite this article
Machado, R.D., Christoff, A.P., Loss-Morais, G. et al. Comprehensive selection of reference genes for quantitative gene expression analysis during seed development in Brassica napus . Plant Cell Rep 34, 1139–1149 (2015). https://doi.org/10.1007/s00299-015-1773-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-015-1773-1