
Abstract
Key message
Wheat genes differentially expressed in the egg cell before and after fertilization were identified. The data support zygotic gene activation before the first cell division in wheat.
Abstract
To have an insight into fertilization-induced gene expression, cDNA libraries have been prepared from isolated wheat egg cells and one-celled zygotes. Two-hundred and twenty-six egg cell and 253 zygote-expressed EST sequences were determined. Most of the represented transcripts were detected in the wheat egg cell or zygote transcriptome at the first time. Expression analysis of fourteen of the identified genes and three controls was carried out by real-time quantitative PCR. The preferential expression of all investigated genes in the female gametophyte-derived samples (egg cells, zygotes, two-celled proembryos, and basal ovule parts with synergids) in comparison to the anthers, and the leaves were verified. Three genes with putative signaling/regulatory functions were expressed at a low level in the egg cell but exhibited increased (2-to-33-fold) relative expression in the zygote and the proembryo. Genes with high EST abundance in cDNA libraries exhibited strong expression in the egg cell and the zygote, while the ones coding for unknown or hypothetical proteins exhibited differential expression patterns with preferential transcript accumulation in egg cells and/or zygotes. The obtained data support the activation of the zygotic genome before the first cell division in wheat.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The life cycle of plants can be divided into a diploid sporophytic and a haploid gametophytic phase. Most of our knowledge on plants comes from dominant sporophytes, because the gametophytes, which consist of only a few cells, are encapsulated in the tissues of the sporophytic generation. Advances in plant molecular biology allowed us to have deeper insights into the plant gametophyte development (for reviews, Yang et al. 2010; Sundaresan and Alandete-Saez 2010; Borg and Twell 2010; Borg et al. 2009).
The female gametophyte of angiosperms typically consists of one egg cell, one central cell, two synergid and three antipodal cells. These cell types all have unique structural features and functions to ensure the success of the reproductive process. Modern, high-throughput techniques allowed studying the transcriptome of these specialized cells in model species (for review, Schmidt et al. 2012). Functional characterization of the transcripts revealed differences highlighting specific post-transcriptional regulatory modules and metabolic pathways characteristic for each female gametophytic cell type (Wuest et al. 2010). However, comparison of transcripts enriched in the egg and synergid cells, respectively, of Arabidopsis and rice also revealed considerable species-specific differences in the molecular networks underlying gametophyte development and function (Ohnishi et al. 2011).
The egg cell has a distinct role in the female gametophyte, because it acts as a signaling center for the development of all female gametophytic cells (Volz et al. 2012) and develops to the new sporophytic generation after fertilization. Wuest et al. (2010) could identify 431 genes which are likely to be specifically expressed in the mature female gametophyte of Arabidopsis thaliana, of which 163 was specifically expressed in the egg. In animals, maternally deposited mRNAs in the egg cell control early embryonic development before the activation of the zygotic genome (Minami et al. 2007). Although, based on various experimental data, a similar period of maternal control on early embryogenesis was hypothesized for plants as well (Baroux et al. 2008), a recent comprehensive study revealed that the zygotic genome is activated right before the first cell division and the earliest phases of embryogenesis are mostly under zygotic control in Arabidopsis (Nodine and Bartel 2012). Comparative gene expression profiling of tobacco egg cells, one- and two-celled zygotes resulted in a similar conclusion (Zhao et al. 2011).
Wheat (Triticum aestivum L.) is one of the most important crops worldwide. Identification of wheat genes and proteins, determining egg-cell development and identity, fertilization success, and early embryogenesis, is of great significance for future practical applications in addition to their scientific value. However, wheat has a huge and complex genome that could not have been fully revealed and, therefore, the adaptation of high-throughput genomic methods for this species is still limited (Gupta et al. 2008). In contrast, there are routine methodologies for the isolation and culture of the cell types of the wheat female gametophyte (Kovács et al. 1994; Kumlehn et al. 1999, 2001). Therefore, the analysis of gene expression profiles of wheat egg cells and zygotes via EST sequencing is feasible.
It was well demonstrated by Sprunck et al. (2005), who prepared cDNA libraries from isolated wheat egg cells and two-celled proembryos. They determined 404 and 789 EST sequences, respectively, and based on the data, compared the expression profiles of the egg cell and the two-celled proembryo. They concluded that the unfertilized wheat egg cell has a higher metabolic activity and protein turnover than previously thought. Moreover, they found that the transcript composition of the proembryos is significantly distinct from that of the egg cells. Transcripts, associated with DNA replication as well as with high transcriptional and translational activity, characterize the transcriptome of the dividing zygote.
More recently, Kőszegi et al. (2011) coupled EST sequencing with a cDNA subtraction approach and identified the egg-cell-specific RKD transcription factors regulating several egg-cell-specific genes.
Here, an EST-sequencing approach is reported using unfertilized wheat egg cells and single-celled zygotes (7 h after fertilization) for cDNA library production and EST sequencing. The obtained data complement of those reported by Sprunck et al. (2005) providing information on single-celled zygote- and further egg-cell-expressed genes of wheat. Furthermore, the reported gene-expression analysis of selected genes supports the activation of zygotic genes before the first cell division in wheat as well.
Materials and methods
Plant material, cultivation and sample collection
Plants of a spring wheat (T. aestivum L.) genotype Siete Cerros were used in the experiment. After germination, plants were vernalized for 5 weeks at 2 °C and transplanted into a sand–soil–peat (1:3:1) mixture (2 kg/pot). Plants were grown in controlled conditions until flowering in a PGR-15 phytotron chamber using the climatic program T2 (Tischner et al. 1997) at a light intensity of 500 μmol m−2 s−1 for 8 weeks. During this period, the initial max/min day/night temperature was increased from 12.5/5.5 to 23/14 °C. Vernalization was applied to spring wheat, because the initial max/min day/night temperature of the T2 climatic program was not sufficient to induce the vegetative/generative transition. The relative day/night humidity of the air circulating in the chamber was 65/75 %. The photosynthetic photon flux density during cultivation was 500 μmol m−2 s−1.
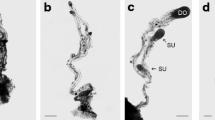
Plants were emasculated, and the spikes were protected with cellophane bags to avoid self- and cross-pollination 4 days before anthesis. Reference spikes were also used to check the efficiency of emasculation and hand pollination, which was above 95 %. Prior to egg cell, zygote, proembryo and ovule isolation, the spikes were surface sterilized with 2 % (v/v) sodium hypochlorite for 20 min and washed four times with sterile distilled water. Egg cells (Fig. 1a, b; n = 50), were isolated from non-pollinated pistils, single-celled zygotes (fertilized egg cells at around the time of karyogamy) (Fig. 1c, d; n = 50), and two-celled proembryos (Fig. 1e, f; n = 50), from hand-pollinated pistils 7 and 24 h after pollination (HAP), respectively, according to the method described earlier by Kovács et al. (1994).

Light and fluorescent micrographs of an unfertilized mature egg cell (a, b), an one-celled zygote (7 h after fertilization; c, d) and a bicellular pro-embryo (24 h after fertilization; e, f)
Basal ovule parts (n = 50), containing the synergids, isolated from non-pollinated, 7 HAP and 24 HAP pistils were collected after egg cell, zygote and proembryo isolation, respectively. Anthers (n = 12) were collected in tricellular stage of pollen development, 1 day before anthesis. All the samples above were collected in 10-10 μl-s of the Lysis/Binding buffer of the “Dynabeads mRNA Direct” RNA isolation kit (Invitrogen, USA) in three repetitions. Leaf samples (3 mg each) were excised from 5-day-old seedlings and placed into RNAlater solution (Invitrogen) in three repetitions. The RNAlater solution was changed for 100 μl Lysis/Binding buffer before RNA isolation.
mRNA isolation and cDNA library preparation
mRNA isolation was carried out according to the protocol provided by the manufacturer (“Dynabeads mRNA Direct” RNA isolation kit; Invitrogen) after adding 90 μl-s of Lysis/Binding buffer and 10 μl oligo-dT-coated Dynabeads to the samples. The mRNA samples have been eluted into 5 μl sterile water and were immediately used in total for cDNA library preparation using the “SMART cDNA Library Construction Kit” (Clonetech Laboratories, USA). First-strand cDNAs have been synthesized by Supercript II reverse-transcriptase (Invitrogen) using the adaptors provided in the kit. Two-stranded cDNA was produced by PCR amplification for 30 cycles due to the limited starting material. This number of cycles still resulted in exponential target amplification as determined experimentally (data not shown). The adaptor-containing cDNAs were digested by Sfi I and ligated to ΛTriplEx2 phagemid arms. Recombinant phages were produced using the Gigapack III Gold Packaging Extract (Stratagen, USA).
EST sequencing
Individual phage plaques were converted to pTriplEx2 plasmids using Escherichia coli BM25.8 as a host as described in the manual of the cDNA library construction kit. Plasmid DNAs were purified using a modified alkali lysis protocol (Feliciello and Chinali 1993). Colony PCR has been carried out using pTriplEx2 5′ sequencing (5′ TCCGAGATCTGGACGAGC 3′) and the T7 promoter (5′ TAATACGACTCACTATAGGGC 3′) primers at 94 °C 5′ 1×, 94 °C 15″ 52 °C 30″ 72 °C 1′ 30″ 28×, 72 °C 5′ 1× PCR cycle parameters using Fermentas (Lithuania) Taq DNA polymerase. Plasmids with inserts >100 bp have been selected for sequencing by Macrogene Inc. (Korea) using the pTriplEx2 5′ sequencing primer. The EST sequences derived from the egg cell were designated as EPS# and those from the zygote as ZIG#. The sequences have been deposited in the EMBL Nucleotide Sequence Database with the accession numbers HE862417–HE862958 (http://www.ebi.ac.uk/embl/).
Sequence analysis and annotation
The homology of vector sequence-free ESTs to database sequences was investigated using the NCBI BLAST server (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequence annotation and annotation analysis were carried out by the BLAST2GO analysis tool (Götz et al. 2008) using default settings.
Real-time quantitative PCR
mRNA isolation and cDNA synthesis were made as described above for cDNA library preparation. For real-time quantitative PCR (RT-QPCR) assays and evaluations, an ABI 7900HT equipment and its software (SDS version 2.3; Applied Biosystems, USA) were used based on SYBR-green detection and ΔΔCT analysis as described elsewhere in more detail (Szűcs et al. 2006). Three assays were carried out on independent samples with at least two parallel measurements each. The RT-QPCR master mix was purchased from Applied Biosystems. The primers used are listed in Supplementary material 1.
Results
The cDNA libraries
The cDNA libraries contain approximately 2.4 × 106 and 0.45 × 106 individual recombinant phages in the cases of unfertilized egg cells and one-celled zygotes, respectively, based on the phage titer and the ratio of insert containing/empty phagemids. The average insert size was determined as 490 bp for egg cell ESTs (EPS) and 416 bp for zygote ESTs (ZIG). The redundancy of the clones was low as the randomly selected 246 egg cell EST sequences represented 226, while the 297 zygote ones 253, different genes (singletons).
Characterization of EST sequences of wheat genes expressed before and/or after fertilization
Randomly selected recombinant phagemids (300-300 from both libraries) have been converted into pTripleEx2 plasmid clones. Plasmids carrying inserts larger than 100 bp have been purified and subjected to sequencing using a 5′ pTriplEx2 vector primer. As a result, 246 EPS and 297 ZIG high-quality sequences could be obtained. The ESTs have been subjected to BLAST analysis and annotation following the in silico removal of vector sequences. Detailed results of these analyses are summarized as Supplementary material 2 (egg cell) and 3 (zygote).
Some of the genes were represented by several ESTs (Table 1) that may indicate abundant expression in the given cell type. However, it has to be taken into account that the cDNA library has been made by SMART PCR. The advantage of using the SMART PCR method is that it preferentially enriches for full-length transcripts. However, in doing so, it may introduce compositional biases and alter the relative abundance of transcripts. In the EPS library, sequences representing TA64547_4565 coding for a putative dihydrolipoamide dehydrogenase were found to be the most redundant (7 occurrences) while in the ZIG library CD889724 coding for a hypothetical protein (12 occurrences).
From the 246 EPS sequences, 208 showed similarity to database sequences based on the BLASTX algorithm (among which 76 was similar to hypothetical or expressed proteins with no known function), and a further 19 using only the BLASTN homology search. The remaining 19 sequences had no homology to known ESTs/genes/proteins. Considering the ZIG ESTs, these numbers were: 297 total; 258 BLASTX (113 hypothetical or expressed proteins); 21 only BLASTN; 18 no homology.
It was also tested how many of the ESTs correspond to transcripts that have already been found to occur in egg cell or zygote cDNA libraries based on the sequences stored in the “Wheat Transcript Assembly 2” dataset from TIGR (The Institute for Genomic Research; updated July 2007). The overlap was minimal (<1 %) indicating that most of the identified EST sequences carry novel information on the expression of genes in wheat egg cells and zygotes.
Selection of genes for expression analysis
Fourteen sequences have been selected for further gene-expression analysis based on the annotation of the represented genes.
Three of the ZIG ESTs were selected because they represent genes with putative roles in signal transduction (ZIG75 was annotated as putative mitogen-activated protein kinase kinase, TaMAPKK) and transcriptional regulation (ZIG43 was annotated as E2F transcription factor-like, TaE2F; ZIG45 was annotated as transcription factor B3-like, TaTFB3). Due to their potential regulatory functions, it was to be investigated whether their transcription is induced by fertilization. The relative expression of the TaSERK3 gene coding for a putative somatic embryogenesis receptor kinase was also determined to serve as a kind of control as the protein was hypothesized to have a role in zygotic embryogenesis (Singla et al. 2008).
Further, four zygote-expressed sequences (ZIG 47,253, 298 303,) were selected based on their unique sequences. As they have no sequence homologues in the available databases, we supposed that at least some of them may have fertilization/zygote specific expression pattern. A similar approach was followed by Kőszegi et al. (2011) who identified in this way the egg-cell-specific members of the wheat RKD transcription factor family.
From the egg-cell library, five ESTs (EPS 47, 49, 104, 124, and 282) were randomly selected from those that represented genes that code for hypothetical proteins or had no homologous sequences in the protein databases as determined by the BLASTX algorithm.
EPS76 and EPS87 sequences were chosen as positive controls as they have several ESTs but only from wheat egg-cell-derived cDNA libraries, therefore, they may code for egg-cell-specific transcripts. The EPS76 represented gene (TaECA1) is homologous to a barley (Hordeum vulgare L.) gene the product of which is annotated as “Early Culture Abundant Protein 1” (HvECA1, gene bank accession: AAF23356.1) as it is expressed in embryogenic microspore cultures during the early culture phase (Vrinten et al. 1999). The gene represented by the EST sequence EPS87 (TaDSUL) codes for a protein with small ubiquitin-like modifier (SUMO) and ubiquitin-like domains (di-SUMO-like or DSUL, gene bank accession: ACL50300.1). Recently, the maize homologue of this protein has been characterized in detail by Srilunchang et al. (2010) who specified that it is required for nuclei positioning, cell specification and viability during female gametophyte maturation.
To have appropriate reference genes expressed ubiquitously in the wheat plant, including the egg cell and zygote, the “Wheat Transcript Assembly 2” dataset from TIGR has been analyzed for genes equally represented by EST sequences in various wheat cDNA libraries (including those made from egg cells or zygotes). Two of the genes, coding for a putative NADPH oxidase (TA61480_4565) and for a hypothetical ubiquitin conjugase (TaUBC; TA64564_4565), identified by this in silico gene-expression approach (for details see Szűcs et al. 2010) were used as reference genes in the RT-QPCR experiments.
Gene-expression analysis
Samples were collected from mature egg cells (at anthesis), one-celled zygotes (7 HAP), proembryos (24 HAP) (see Fig. 1), and from the lower ovule parts, they were removed off (at anthesis, 7 and 24 HAP), as well as from anthers and young leaves.
As it is shown in Fig. 2, the relative expression of the four investigated genes annotated as having potential functions in signaling/transcription (TaMAPKK, TaE2F, TaTFB3, TaSERK3) was lower in leaves or anthers in comparison to the ovule-derived samples. Three of them exhibited an increase in their relative mRNA level in response to the fertilization (in the zygote/proembryo vs. the egg cell). This increase was strong (more than 20- and 5-fold, respectively) in the case of the two transcription factors TaTFB3 and TaE2F. The relative expression of the TaSERK3 gene was the highest in the ovules. Its expression was increased by more than twofold in the fertilized egg cell and the 1-day-old zygote as compared to the unfertilized egg cell. The TaMAPKK gene showed a rather constitutive expression in these samples. As far as the relative expression of these genes in the egg cell is considered, the TaMAPKK gene exhibited a high relative expression (similar to the expression level of the reference gene coding for an ubiquitin conjugase) while the transcripts of the three other genes were much less abundant (approximately two orders of magnitude lower abundance).

The relative expression of genes selected based on their potential signaling/regulatory role during fertilization. a The relative expression of the genes in various cell types/organs. Expression in the egg cell was chosen as a reference (relative expression = 1). b Relative expression of the genes in the egg cell. The TaUBC gene expression was chosen as a reference (relative expression = 1). TaSERK3 was used as a positive control (Singla et al. 2008). Expression in the egg cell was normalized to the expression of the genes coding for a NADPH oxidase (TA61480_4565) and an ubiquitin conjugase (TA64564_4565). LEAF young leaf; ANT anther; OV basal ovule part with synergids; the numbers indicate the time of isolation after fertilization in hours; EC unfertilized egg cell, ZYG zygote, isolated 7 h after fertilization; ProE bicellular proembryo, isolated 24 h after fertilization. Average of relative gene expression values derived from three independent sampling, RNA isolation and cDNA synthesis is shown with standard errors
The nine genes represented by ESTs with no homology to known sequences and the two control ones (TaECA1; TaDSUL) all exhibited strong egg-cell/zygote/proembryo-specific relative expression as compared to ovule-parts, anthers and leaves. These genes were classified into four categories based on their expression pattern. TaDSUL, TaECA1, EPS49, EPS104, and ZIG298 exhibited decreased expression in the proembryo in comparison to the egg cell and zygote where they were expressed at more or less the same level (Fig. 3a). EPS124 and ZIG47 genes exhibited a rather constitutive expression in these cell types (Fig. 3b). EPS47 and ZIG253 showed a moderately (2-to-3-fold) increased zygotic expression (Fig. 3c), while EPS282 and ZIG303 showed a high increase in their relative mRNA levels in the zygote and proembryo as compared to the egg cell (Fig. 3d).

The relative expression of genes represented by ESTs with no established homology to proteins with known function. a–d The relative expression of the genes in various cell types/organs. Expression in the egg cell was chosen as a reference (relative expression = 1). Genes are shown in separate histograms according to their expression pattern. e Relative expression of the same genes in the egg cell. The TaUBC gene expression was chosen as a reference (relative expression = 1). TaECA1 (homologous to HvECA1, Vrinten et al. 1999) and TaDSUL (Srilunchang et al. 2010) genes were used as positive controls for egg cell/zygote-specific expression. Expression in the egg cell was normalized to the expression of the genes coding for a NADPH oxidase (TA61480_4565) and an ubiquitin conjugase (TA64564_4565). LEAF young leaf; ANT anther; OV basal ovule part with synergids; the numbers indicate the time of isolation after fertilization in hours; EC unfertilized egg cell; ZYG zygote, isolated 7 h after fertilization; ProE two-celled proembryo, isolated 24 h after fertilization. Average of relative gene expression values derived from three independent sampling, RNA isolation and cDNA synthesis is shown with standard errors
The normalized mRNA levels of the selected genes were compared in the egg cell to have a view on their relative expression strength in this cell type (Fig. 3e). Based on this parameter, the genes could be classified into three categories. The EPS47 gene exhibited a rather low expression level in the egg cell. In contrast, the two genes with egg-cell-specific in silico expression pattern (TaECA1 and TaDSUL) as well as another gene represented by EPS104 exhibited a strong relative expression in this cell type. The further seven investigated genes showed an “intermediate” expression level as compared to the two above groups.
Discussion
Egg cells of angiosperms develop from a single megaspore mother cell through a series of meiotic and mitotic divisions together with the three other cell types of the female gametophyte, the synergids, the antipodal cells and the central cell. Unfolding of developmental pathways leading toward the formation of the female gametophyte, including pattern formation and cell specification, recently achieved considerable progress due to studies of Arabidopsis mutants (for reviews, Sundaresan and Alandete-Saez 2010; Yang et al. 2010). However, the unique features of the egg cell underlying its biochemical identity, metabolic activity, and developmental potential are still poorly understood (Russell 1993). Recent genomic approaches allowed a deep insight into the egg-cell transcriptome of the model species Arabidopsis and rice (Wuest et al. 2010; Ohnishi et al. 2011) and these data indicate considerable species-specific differences.
Here, the production of a representative phagemid cDNA library prepared from isolated wheat egg cells (and from one-celled zygotes; see further) is reported. The low redundancy of the library allowed the identification of more than 200 new wheat egg-cell transcribed genes via EST sequencing (see Supplementary material 2 for the detailed gene list).
The EST sequence population generated in this study hardly have any overlap with those that have been previously deposited in public databases by others using similar approaches. A possible explanation is the use of different methods for cDNA library preparation and sequencing. The SMART cDNA Library technology applied in the present study resulted in the enrichment of full-length cDNA sequences, which are cloned directionally into the pTriplEx2 phagemid vector. As we used the 5′ pTriplEx2 sequencing primer for EST generation, our data represent 5′ non-coding and coding sequences underrepresented in other EST populations generated from unidirectionally cloned cDNA fragments (e.g., Sprunck et al. 2005). In the absence of the wheat genome sequence, it cannot be excluded, however, that the same transcripts are represented by different non-overlapping ESTs in the various studies.
The EST-sequencing approach was also suitable for the identification of transcripts with preferential accumulation in the egg cell. As the result of a similar study, Sprunck et al. (2005) have reported several genes to be expressed specifically in the wheat egg cell and two-celled zygote (proembryo) in comparison to vegetative cells. More recently, Kőszegi et al. (2011) reported on the identification of wheat egg-cell-expressed transcription factors belonging to the plant-specific RKD family. The ectopic expression of the Arabidopsis homologues of these transcription factors induced proliferation and the expression of egg-cell-specific genes in vegetative tissues (Kőszegi et al. 2011). These previous studies clearly indicated the potentials of the EST-sequencing approach in cell-specific transcript identification. We could extend the wheat female gametophyte-specific gene set via analyzing the expression of nine egg-cell and zygote ESTs that code for unknown or hypothetical proteins. The assumption that the under-representation of these sequences in the databases is due to their cell/development-specific expression pattern was validated by RT-QPCR analysis.
Fertilization, the fusion of female and male gametes of sexually reproducing multicellular organisms initiates a series of events (maternal-to-zygotic transition, MZT) that finally leads to the development of a new organism through embryogenesis. The timing of zygotic gene activation (ZGA) in plants is somewhat controversial (Baroux et al. 2008; Zhao et al. 2011; Nodine and Bartel 2012). Recent data, however, indicate that it follows fertilization very early and the first steps of embryogenesis are mostly under zygotic control in Arabidopsis and tobacco (Nodine and Bartel 2012; Zhao et al. 2011). In animals, the extent of maternal control and the timing of MZT vary greatly among species, and the MZT not always coincides with the gradual process of ZGA (for review; Shen-Orr et al. 2010). The variability in the timing and the molecular background of ZGA and MZT among plant species is currently unknown.
Sprunck et al. (2005) compared a set of ESTs from 1-day-old two-celled wheat zygotes to that of mature egg cells. Their EST-sequencing data, complemented by the expression analysis of selected genes, indicated that the zygotic genome is already activated in the proembryos. In the present work, we complemented these data selecting an earlier time point (7 HAP) for the isolation of zygotes. It was supposed that at this rather early time point, the direct effect of the fertilization event on the zygotic transcriptome can be investigated including the activation of genes associated with the cell-cycle entry. Moreover, in this way, we could also define whether ZGA precedes the first zygotic cell division in wheat. The increased relative expression of several genes in the single-celled wheat zygote in comparison to the egg cell indeed indicated a very early ZGA in wheat. Similar assumption was obtained by the comparison of gene expression in tobacco egg cells and one-celled zygotes using cDNA subtraction (Ning et al. 2006) and EST-sequencing (Zhao et al. 2011) approaches. Therefore, Arabidopsis, tobacco and wheat data support that ZGA starts in plants before the first cell division of the zygote.
Although in the present study only a relatively low number of egg cell and zygotic ESTs were sequenced (less than 300 each), the transcripts related to signaling (e.g. MAPKK), cell cycle progression (E2F transcription factor, Cullin, Skp1, etc.) or transcription (A2 and B3 domain transcription factors, etc.) were enriched among the zygotic ones (see the whole gene lists as Supplementary materials 2 and 3). The expression of three of these genes (MAPKK, E2F, and TFB3) was analyzed in more detail. MAP-kinase cascades are central to mitogen signaling in eukaryotes, including plants (Mishra et al. 2006), and the role of specific MAPKKKs (upstream regulators of MAPKKs) in the post-fertilization signaling events of Solanum chacoense ovules has already been demonstrated (Gray-Mitsumune et al. 2006). Although we could not see a strong induction in the expression of TaMAPKK, one can suppose that the activity of this kinase is regulated post-translationally in response to fertilization.
The low relative expression level of the transcription factors E2F and TFB3 in the egg cell was found to be considerably augmented in response to fertilization. It has to be emphasized that E2F transcription factors are important regulators of the cell cycle (especially at the G0/G1-S cell-cycle phase transition; Berckmans and De Veylder 2009); several B3-domain transcription factors have been associated with the initiation and progression of embryogenesis (Suzuki and McCarty 2008). Therefore, a similar role for TaE2F in the first zygotic cell division, and TaTFB3 in embryogenesis, may also be hypothesized based on their sequence homology and expression pattern. In contrast, the relative expression of the TaSERK3 gene previously hypothesized to be associated with embryogenesis (Singla et al. 2008) was lower in the egg cell, zygote and proembryo as compared to the ovule from where these cells have been removed. This indicates a more general role for this kinase in the gametophyte development.
The preferentially egg-cell-specific expression of TaECA1 and TaDSUL having their sequences abundantly represented among wheat egg cell ESTs (Sprunck et al. 2005 and the present study) was confirmed by RT-QPCR analysis. The maize homologue of the TaDSUL protein may be involved in the post-translational regulation of other proteins by sumoylation during female gametophyte maturation (Srilunchang et al. 2010). This gene was reported to be exclusively expressed in the micropylar region of the immature female gametophyte, while after cellularization, its expression was restricted to the egg cell and the zygote (Srilunchang et al. 2010). The transcripts coding for HvECA1-homologous proteins were found to be the most abundant transcripts in tobacco as well as wheat egg cells as well (Sprunck et al. 2005; Ning et al. 2006; Zhao et al. 2011). The biochemical or molecular functions of ECA1 homologues are, however, not known yet. The EPS104 sequence with no annotation represents a gene with similar expression properties. Therefore, these genes are good candidates to isolate strong egg-cell-specific plant promoters that could potentially be used to manipulate female fertility and/or parthenogenesis.
In addition to the annotated sequences, we have analyzed the expression of several ESTs that code for hypothetical or unknown proteins. If the sequence/function of the represented genes will be identified, e.g., following the sequencing of the whole wheat genome, it will contribute to our knowledge about the molecular and cellular events underlying the first steps of zygotic embryogenesis in plants.
The EST-sequencing approaches have inherent drawbacks as they are relatively low throughput, expensive and generally not quantitative. Establishing the exact inventory and timing of fertilization-induced events in plants may be facilitated by the recent progress in high throughput, quantitative, and cheaper RNA-sequencing approaches (Schmidt et al. 2012; Schmid et al. 2012). The state-of-the-art and the future potential of using these new techniques in the analysis of plant gametophyte development have recently been reviewed by Schmid et al. (2012).
References
Baroux C, Autran D, Gillmor CS, Grimanelli D, Grossniklaus U (2008) The maternal to zygotic transition in animals and plants. Cold Spring Harb Symp Quant Biol 73:89–100
Berckmans B, De Veylder L (2009) Transcriptional control of the cell cycle. Curr Opin Plant Biol 12:599–605
Borg M, Twell D (2010) Life after meiosis: patterning the angiosperm male gametophyte. Biochem Soc Trans 38:577–582
Borg M, Brownfield L, Twell D (2009) Male gametophyte development: a molecular perspective. J Exp Bot 60:1465–1478
Feliciello I, Chinali G (1993) A modified alkaline lysis method for the preparation of highly purified plasmid DNA from Escherichia coli. Anal Biochem 212:394–401
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Gray-Mitsumune M, O’Brien M, Bertrand C, Tebbji F, Nantel A, Matton DP (2006) Loss of ovule identity induced by overexpression of the fertilization-related kinase 2 (ScFRK2), a MAPKKK from Solanum chacoense. J Exp Bot 57:4171–4187
Gupta PK, Mir RR, Mohan A, Kumar J (2008) Wheat genomics: present status and future prospects. Int J Plant Genomics 2008:896451
Kőszegi D, Johnston AJ, Rutten T, Czihal A, Altschmied L, Kumlehn J, Wüst SEJ, Kirioukhova O, Gheyselinck J, Grossniklaus U, Bäumlein H (2011) Members of the RKD transcription factor family induce an egg cell-like gene expression program. Plant J 67:280–291
Kovács M, Barnabás B, Kranz E (1994) The isolation of viable egg cells of wheat (Triticum aestivum L.). Sex Plant Reprod 7:311–312
Kumlehn J, Lörz H, Kranz E (1999) Monitoring individual development of isolated wheat zygotes: a novel approach to study early embryogenesis. Protoplasma 208:156–162
Kumlehn J, Kirik V, Czihal A, Altschmied L, Matzk F, Lörz H, Bäumlein H (2001) Parthenogenetic egg cells of wheat: cellular and molecular studies. Sex Plant Reprod 14:239–243
Minami N, Suzuki T, Tsukamoto S (2007) Zygotic gene activation and maternal factors in mammals. J Reprod Dev 53:707–715
Mishra NS, Tuteja R, Tuteja N (2006) Signaling through MAP kinase networks in plants. Arch Biochem Biophys 452:55–68
Ning J, Peng XB, Qu LH, Xin HP, Yan TT, Sun M (2006) Differential gene expression in egg cells and zygotes suggests that the transcriptome is restructed before the first zygotic division in tobacco. FEBS Lett 580:1747–1752
Nodine MD, Bartel DP (2012) Maternal and paternal genomes contribute equally to the transcriptome of early plant embryos. Nature 482:94–97
Ohnishi T, Takanashi H, Mogi M, Takahashi H, Kikuchi S, Yano K, Okamoto T, Fujita M, Kurata N, Tsutsumi N (2011) Distinct gene expression profiles in egg and synergid cells of rice as revealed by cell type-specific microarrays. Plant Physiol 155:881–891
Russell SD (1993) The egg cell: development and role in fertilization and early embryogenesis. Plant Cell 5:1349–1359
Schmid MW, Schmidt A, Klostermeier UC, Barann M, Rosenstiel P, Grossniklaus U (2012) A powerful method for transcriptional profiling of specific cell types in eukaryotes: laser-assisted microdissection and RNA sequencing. PLoS ONE 7:e29685
Schmidt A, Schmid MW, Grossniklaus U (2012) Analysis of plant germline development by high-throughput RNA profiling: technical advances and new insights. Plant J 70:18–29
Shen-Orr SS, Pilpel Y, Hunter CP (2010) Composition and regulation of maternal and zygotic transcriptomes reflects species-specific reproductive mode. Genome Biol 11:R58–R58
Singla B, Khurana JP, Khurana P (2008) Characterization of three somatic embryogenesis receptor kinase genes from wheat, Triticum aestivum. Plant Cell Rep 27:833–843
Sprunck S, Baumann U, Edwards K, Langridge P, Dresselhaus T (2005) The transcript composition of egg cells changes significantly following fertilization in wheat (Triticum aestivum L.). Plant J 41:660–672
Srilunchang KO, Krohn NG, Dresselhaus T (2010) DiSUMO-like DSUL is required for nuclei positioning, cell specification and viability during female gametophyte maturation in maize. Development 137:333–345
Sundaresan V, Alandete-Saez M (2010) Pattern formation in miniature: the female gametophyte of flowering plants. Development 137:179–189
Suzuki M, McCarty DR (2008) Functional symmetry of the B3 network controlling seed development. Curr Opin Plant Biol 11:548–553
Szűcs A, Dorjgotov D, Otvös K, Fodor C, Domoki M, Györgyey J, Kaló P, Kiss GB, Dudits D, Fehér A (2006) Characterization of three Rop GTPase genes of alfalfa (Medicago sativa L.). Biochim Biophys Acta 1759:108–115
Szűcs A, Jäger K, Jurca ME, Fábián A, Bottka S, Zvara A, Barnabás B, Fehér A (2010) Histological and microarray analysis of the direct effect of water shortage alone or combined with heat on early grain development in wheat (Triticum aestivum). Physiol Plant 140:174–188
Tischner T, Köszegi B, Veisz O (1997) Climatic programs used in the Martonvásár phytotron most frequently in recent years. Acta Agron Hung 45:85–104
Volz R, von Lyncker L, Baumann N, Dresselhaus T, Sprunck S, Gross-Hardt R (2012) LACHESIS-dependent egg-cell signaling regulates the development of female gametophytic cells. Development 139:498–502
Vrinten PL, Nakamura T, Kasha KJ (1999) Characterization of cDNAs expressed in the early stages of microspore embryogenesis in barley (Hordeum vulgare) L. Plant Mol Biol 41:455–463
Wuest SE, Vijverberg K, Schmidt A, Weiss M, Gheyselinck J, Lohr M, Wellmer F, Rahnenführer J, von Mering C, Grossniklaus U (2010) Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr Biol 20:506–512
Yang WC, Shi DQ, Chen YH (2010) Female gametophyte development in flowering plants. Annu Rev Plant Biol 61:89–108
Zhao J, Xin H, Qu L, Ning J, Peng X, Yan T, Ma L, Li S, Sun MX (2011) Dynamic changes of transcript profiles after fertilization are associated with de novo transcription and maternal elimination in tobacco zygote, and mark the onset of the maternal-to-zygotic transition. Plant J 65:131–145
Acknowledgments
The presented work was supported by grants (K-67987 and K-68064) of the Hungarian Scientific Research Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W. Harwood.
M. Domoki and A. Szűcs contributed equally to the research.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Domoki, M., Szűcs, A., Jäger, K. et al. Identification of genes preferentially expressed in wheat egg cells and zygotes. Plant Cell Rep 32, 339–348 (2013). https://doi.org/10.1007/s00299-012-1367-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-012-1367-0