
Abstract
Transposable elements (TEs) dominate the genetic capacity of most eukaryotes, especially plants, where they may compose up to 90% of the genome. Many studies, both in plants and animals reported that in fact non-autonomous elements that have lost their protein-coding sequences and became miniature elements were highly associated with genes, and showed a high level of transpositional activity such as mPing family in rice. In this study, we have investigated in detail the copy number, insertional polymorphism and the methylation status of the tiniest LTR retrotransposon family, termed TRIM, in nine rice strains, in comparison with mPing. While TRIM showed similar copy numbers (average of 79 insertions) in all the nine rice strains, the copy number of mPing varied dramatically (ranging from 6 to 203 insertions) in the same strains. Site-specific PCR analysis revealed that ~58% of the TRIM elements have identical insertion sites among the nine rice strains, while none of the mPing elements (100% polymorphism) have identical insertion sites in the same strains. Finally, over 65% of the TRIM insertion sites were cytosine methylated in all nine rice strains, while the level of the methylated mPing insertion sites ranged between 43 and 81.5%. The findings of this study indicate that unlike mPing, TRIM is most probably a fossil TE family in rice. In addition, the data shows that there might be a strong correlation between TE methylation and copy number.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) are considered one of the most important genomic components that might play a prominent role in organismal biology because: first, TEs makeup a large fraction of most eukaryotic genomes, particularly grasses, where they account for up to 90% of the genome (Sabot et al. 2005), and second, the massive variation in TE content and activity among species. Transposable elements are DNA fragments that are able to move from one location in the genome to another, either by a “copy and paste” mechanism (class I), or by a “cut and paste” mechanism (class II). The movement of class I elements is mediated by the production of RNA intermediates, while class II elements move via DNA intermediates (Wicker et al. 2007). Because of the “copy and paste” nature of class I elements, they are able to attain enormous copy numbers (up to a million copies, such as Alu elements in humans (Xing et al. 2009)). Usually, TEs are considered “selfish” or “parasitic” because their proliferation is negatively correlated with the fitness of their host (Slotkin and Martienssen 2007).
Transposable element transposition can cause various mutations, such as deletions, insertions, translocations (Slotkin and Martienssen 2007), and can also influence gene expression (Kashkush et al. 2003; Iida et al. 2004; Lockton and Gaut 2009). Usually in plants, a large fraction of TE sequences are targeted for methylation (Kumar and Bennetzen 1999; Rabinowicz et al. 2003; Madlung and Comai 2004), as such TEs are considered epigenetically silenced (Slotkin and Martienssen 2007). For example, most rice TEs show over 50% methylation (Kashkush and Khasdan 2007; Kishima et al. 2007), while other genomic sequences show ~16% methylation (Xiong et al. 1999). In addition, the entire sequence of Arabidopsis TEs is usually methylated in all sequence contexts (Gehring and Henikoff 2008). However, Arabidopsis TEs can be reactivated in genetic backgrounds containing methylation-defective mutants (ddm1) or during tissue culture (Miura et al. 2001; Singer et al. 2001). In a recent study, it was reported that the transpositional activity of a rice miniature inverted-repeat transposable element (MITE) termed mPing was induced following tissue culture treatment, and that this activation was correlated with methylation (Ngezahayo et al. 2009).
Surprisingly, some of the most recently active TEs in both plants and animals are in fact non-autonomous elements (both class I and class II) that have lost their protein-coding sequences and became miniature elements, such as the class I terminal-repeat retrotransposons in miniature (TRIMs) (Witte et al. 2001; Sabot et al. 2005) and the class II miniature inverted-repeat transposable elements (MITEs) (Jiang et al. 2004; Yang et al. 2009).
Miniature inverted-repeat transposable elements are widespread in eukaryotic genomes; they are non-autonomous elements that are characterized by their relatively short sequence, structural similarity, conserved terminal repeats, and high copy number. In plants, most MITEs are classified into two main superfamilies: Tourist-like, and Stowaway-like (Jiang et al. 2004; Feschotte and Pritham 2007). Similarly, TRIMs possess the classical structure of LTR retrotransposons, but they are distinguished by their short sequence (Witte et al. 2001; Sabot et al. 2005). As TRIMs contain poly-purine tract (PPT) and primer binding site (PBS) sequences, they are capable of transposing if the retrotransposition proteins are available from another source. In addition, the high conservation of the TRIMs terminal direct repeats (TDRs) in plants might indicate they retain retrotransposition activity (Witte et al. 2001).
Because of the very high activity that was observed for mPing, we aimed to study the activity of the tiniest class I TRIM family in rice, and to compare its activity with that of the tiniest class II family—mPing. The complete sequence for Oryza sativa ssp. japonica facilitated the design of experiments to assess the copy-number variation (CNV), the insertional polymorphism and the methylation status of both mPing and TRIM in various rice strains.
Materials and methods
Plant material
In this study, nine rice strains originated from different countries were used, including eight O. sativa strains: (1) Plant ID Aikoku Ibaragi 2—PI 637582 (Japan), (2) Plant ID Mubo Aikoku—GSOR 310930 (Japan), (3) Plant ID Ginbozu—PI 388459 (Japan), (4) Plant ID Gimbozu—Clor 6873 (Japan), (5) Plant ID Nipponbare—PI 514663 (Japan); (6) Plant ID Indica16—PI 645480 (Arkansas, USA), (7) Plant ID O-68-07—PI 342917 (India), and (8) Plant ID Nivara 07—PI 431320 (Philippines), and one Oryza officinalis, Plant ID IRRI-IRGC-101073—PI 590412 (Philippines). Seed material was kindly provided by the United States Department of Agriculture (http://www.ars-grin.gov/npgs/acc/acc_queries.html).
Computer-assisted analysis
Retrieving TE sequences from databases
TRIM and MITE sequences were retrieved using the tree analysis of related genes and transposons (TARGeT) web-based pipeline (see details in Han et al. 2009; http://target.iplantcollaborative.org). TARGeT automatically identifies and retrieves homologues sequences to the query input from a certain selected sequenced genome. In this study, we have retrieved TRIM and MITE sequences, together with 1 kb-flanking host DNA sequence from both sides of each retrieved insertion, from the sequence draft of the two O. sativa subspecies, japonica and indica, using default criteria of e-value 0.01, and minimal match percentage (MMP) 70%.
Sequence annotation of TE-flanking sequences relied on the BLAST 2.0 package from NCBI (http://www.ncbi.nlm.nih.gov/BLAST/) and from the Institute for Genomic Research (http://tigrblast.tigr.org/tgi/). No significant sequence hits in databases at e-value <e−10.
Biodiversity analysis
Hierarchical agglomerative clustering analysis of the data with Bray-Curtis similarity and construction of the dendrogram was performed using the Primer6 software version 6.1.6 (Primer-E; (Clarke 1993)). Bands matrix was constructed by designating a PCR product with an expected size for the full site as 1 and an empty site as 0. The similarity profile (SIMPROF) test was used on each node to assess the statistical significance of the dendrogram. SIMPROF calculates a mean profile by randomizing each variable’s values and re-calculating the profile. The pi statistic is calculated as the deviation of the actual resemblance profile of the resemblance matrix with the mean profile. This is compared with the deviation of further randomly generated profiles to test for significance.
Statistical analysis
A correlation analysis was performed using the SAS-based software, JMP 5 (SAS Institute Inc 1995), with standard parameters and at α = 0.05.
Site-specific PCR
For PCR analysis, genomic DNA was isolated from young leaves (age 4 weeks post-germination) using the DNeasy plant kit (QIAGEN). The insertional polymorphism of both TRIMs and MITEs in the nine rice genomes was determined using site-specific PCR (ssPCR) assay. Primers were designed in TE-flanking sequences based on the Nipponbare sequence draft using the Primer3 software version 0.4.0 (http://frodo.wi.mit.edu/primer3/). Primer sequences and expected product sizes are available in Supplemental Table 1. Each PCR reaction contained: 2.0 μl Taq DNA polymerase buffer 10× (Fisher Biotec), 2.0 μl of 25 mM MgCl2 (Fisher Biotec), 0.8 μl 2.5 mM dNTPs, 0.2 μl of Taq DNA polymerase (5 U/μl, Fisher Biotec), 1 μl of forward primer (50 ng/μl), 1 μl of reverse primer (50 ng/μl), 1 μl genomic DNA (30 ng/μl) and 12 μl of ultra pure water (Biological Industries). The final volume of each reaction was 20 μl. PCR conditions included: 94°C for 5 min, repeat 30× (94°C for 1 min, 60°C for 1 min, 72°C for 1 min) and 72°C for 5 min. PCR products were loaded onto 1% agarose gels, and then the gels were stained with ethidium bromide (Amresco) and product size was determined against a 100 bp ladder DNA standard (GeneDire). For sequence validation, PCR products were purified using the Invisorb® Spin PCRapid Kit (Invitek) or extracted from the agarose gel by using the MinElute® Gel Extraction Kit (QIAGEN). Sequencing was done using the 3730 DNA Analyzer (Applied Biosystems) at Ben-Gurion University.
Real-time quantitative PCR
Copy-number variation of TRIM and mPing in every genome was determined by quantitative real-time PCR. Each reaction contained: 7.5 μl of KAPA SYBR® FAST qPCR Master Mix (2×), 0.3 μl ROX Low 50× (KAPA BIOSYSTEMS), 1 μl of forward primer (10 μM), 1 μl of reverse primer (10 μM), 0.2 μl of ultra pure water (Biological Industries) and 5 μl of template genomic DNA (0.4 ng/μl). Primers were designed with Primer Express software version 3.0 (Applied Biosystems). Primer sequences are available in supplemental Table 2. Actin served as endogenous control (Fukao et al. 2011). The qPCR reaction was conducted and analyzed by a 7500 Fast Real-Time PCR system and 7500 Software version 2.0.5 (Applied Biosystems).
Copy-number variation of TRIM and mPing was determined by examining the relative quantity of these elements in the different rice genomes. These relative quantities were calculated according to (Kraitshtein et al. 2010). The Nipponbare genome served as reference genome, therefore its relative quantity was considered as 1 for both TRIM and mPing CNV experiments. In brief, a comparative 2−ΔΔCt method for determining a relative target quantity in samples was used in the normalization and analysis of the relative quantities of both TRIM and mPing. The quantities of target (TRIM or mPing) and the endogenous control (Actin) were measured in samples and in a reference sample (Nipponbare). Then, using 7500 Software version 2.0.5, the relative quantity of target in each sample was determined by comparing the normalized target quantity in each sample to the normalized target quantity in the reference sample, based on the following equation: ΔΔCt(test sample) = [Ct(target) − Ct(Actin)]test sample − [Ct(target) − Ct(Actin)]Nipponbare. Therefore, RQ = (the fold of template amplification at each cycle)−ΔΔC.
Reproducibility of the results was tested by using three technical and three biological replicates for each one of the rice strains. To distinguish specific from non-specific PCR products, a melting curve was generated immediately after amplification. It consisted of 15 s incubation at 95°C and 1 min incubation at 60°C, after which time the temperature was increased by increments of 0.1°C/s until 95°C was reached. The same specific product was detected for either target or reference genes, while no amplification was detected in the no-template control wells.
PCR efficiencies of the target and reference genes were determined by generating standard curves, based on serial dilutions prepared from DNA templates. Fold amplification at each cycle was calculated according to PCR efficiency, which was deduced by the software from the slope of the regression line (y) according to the equation E = [(10−1/y) − 1] × 100. For primers with 100% efficiency, the fold equals 2. For other efficiencies, the software adjusts the fold accordingly (see Supplemental Table 2). Note that quality control for qPCR experiments to rule out possible competition effects in the PCR reactions using template mix was also performed (See supplemental Figure 1).
Transposon methylation display
Methylation levels of CCGG sites flanking TRIMs and mPings in each one of the nine rice strains were tested by a transposon methylation display (TMD) assay (Kashkush and Khasdan 2007). Transposon methylation display allows the analysis of cytosine methylation in CG and CNG contexts at TE-flanking sites. Primer sequences are available in supplemental Table 3. A TE-specific primer from the 5′-terminus (Supplemental Table 3) was used in the TMD together with an adapter primer (Supplemental Table 3).
Note that primers used for mPing were according to (Jiang et al. 2003). Primers were fluorescently labeled and the data was analyzed by GeneMapper version 4 (Applied Biosystems). In brief, DNA was cleaved with HpaII and MspI restriction enzymes (isoschizomers) that recognize CCGG sites, while HpaII is sensitive to methylation of either cytosine (except when the external cytosine is hemi-methylated, i.e., methylation of one DNA strand), MspI is sensitive only when the external cytosine is methylated (both at homo- or hemi-methylation status). Thus, the different types of methylation of CCGG sites resulted in different cleavage products (amplfied by PCR) by the isoschizomers. In case of non-methylated CCGG site, both isoschizomers will produce the same cleavage product. Each TMD band contains a chimeric (TE/flanking DNA) sequence. Note that in some cases TE-internal sequence might also be amplified, thus enabling the analysis of the methylation status in CCGG sites within the transposon.
Results and discussion
In silico characterization of TRIM and MITE elements from the rice sequence draft
TRIM- and MITE-containing sequences were retrieved from the complete sequence of O. sativa ssp. japonica (cv. Nipponbare) using TARGeT software (Han et al. 2009). Overall, 79 intact TRIM elements and 51 mPing elements were retrieved and analyzed. All elements were distributed over the 12 rice chromosomes, with 55% of the TRIM and 54.5% of the MITE elements inserted into or near genes (see Supplemental Tables 4 and 5, respectively). The distribution, structure, conservation, and activity of the mPing elements were analyzed previously (Jiang et al. 2004; Naito et al. 2006) in detail in O. sativa ssp. japonica and were found to be well conserved (all 51 japonica elements are nearly identical in sequence and length, 430 bp) and highly active (Jiang et al. 2003; Kikuchi et al. 2003; Nakazaki et al. 2003; Shan et al. 2005; Naito et al. 2006, 2009; Ngezahayo et al. 2009). In contrast, here we found that TRIMs showed high divergence in their length (ranging in size from 228 to 585 bp), while most TRIMs were ~376 bp long with 115 bp TDRs. While the TDRs of all 79 elements showed ~90% sequence similarity, the internal non-coding sequence showed only ~20% sequence similarity (Supplemental Figure 2). In addition, clear 5-bp target site duplication (TSD) was observed for only 40% of the 79 intact TRIM elements with no significant target site preference (Supplemental Figure 3). Note that we successfully retrieved four elements that we termed long-TRIMs. Two of them are ~5 kb long and the other two are ~1.6 kb. One of the 5 kb long-TRIMs contained a Copia-type Pol-like coding sequence (Fig. 1). The duplicated 26 bp sequence in direct orientation, flanking the new unique sequence of this Copia-type long form indicates that most probably the long form of TRIM was generated as a result of illegitimate integration or a recombination between the short TRIM form and another Copia-like unique sequence (see Fig. 1). A similar phenomenon was seen in the wheat TRIM family termed Veju (Sabot et al. 2005). Another possibility is that the short form of TRIM was generated as a result of a recombination between the two direct 26 bp sequences followed by a deletion of the internal sequence.

Schematic presentation of the long (upper) and the short (lower) forms of TRIM in rice. The identical sequences are indicated (terminal direct repeats-TDRs, and part of the internal non-coding sequence). 26-bp direct repeat in the breakpoints is indicated
The conservation of the mPing family in japonica led to the hypothesis that this element might retain activity in rice. Later it was shown in several studies that the mPing family is the most active DNA transposon in rice, and it has amplified its copy number by hundreds of copies in some sativa strains (Naito et al. 2006). In addition, it was shown that some mPing insertions were associated with the expression of the adjacent genes (Naito et al. 2009). Similarly, because of the high conservation of the TDRs of TRIM insertions and because 55% of the elements are inserted into or near genes, it was speculated (Witte et al. 2001) that TRIMs might be one of the most active retrotransposons in rice. For these reasons, we focused our investigation on the copy-number variation, the insertional polymorphism, and the methylation status of TRIMs in nine rice strains, and those parameters were compared to that of mPing. These analyses will allow us to compare the potential activity of the tiniest class I and class II TE families in rice, respectively.
Copy-number variation (CNV) of TRIMs and MITEs in rice strains
The sequence drafts for japonica and for indica revealed that there are 51 copies of mPing in japonica, and eight copies in indica (Naito et al. 2006). TRIMs, on the other hand, appear in ~79 copies in japonica and ~70 copies in indica. Copy-number variation is one of the important factors that might indicate TE proliferation throughout evolution, thus we assessed the copy number of both mPing and TRIM in nine rice strains (see plant material) using real-time quantitative PCR (Kraitshtein et al. 2010). The qPCR allowed us to measure the relative quantity (RQ) of each TE family in each one of the rice strains, and then the RQ values were converted to actual copy numbers using the copy numbers in japonica as reference (51 mPings and 79 TRIMs). The qPCR experiments had three biological replicates (see “Materials and methods”, Quantitative PCR). Note that quality control for qPCR experiments, to rule out possible competition effects in the PCR reactions using template mix, was also performed (see Supplemental Figure 1).
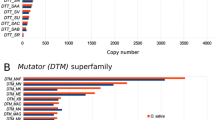
While mPing showed a significant CNV in the eight O. sativa strains: Aikoku Ibaragi 2 (Japan), Mubo Aikoku (Japan), Ginbozu (Japan), Gimbozu (Japan), Nipponbare (Japan), Indica16 (Arkansas, USA), O-68-07 (India), Nivara 07 (Philippines), and in O. officinalis (Philippines) (Fig. 2a), TRIM showed a minor CNV in the same strains (Fig. 2b). The copy number of mPing varies from six copies in Ginbozu to 203 copies in Mubo Aikoku (See Fig. 2a). A greater CNV of mPing was shown in irradiated Gimbozu and Aikoku strains (tens to over 1,000 copies) by (Naito et al. 2006). This is an indication that mPing retain activity in some rice strains (Naito et al. 2006), and perhaps it was active during the ancient and recent evolution of rice. In contrast, the copy number of TRIM varies from 65 copies in O. officinalis to 91 copies in Nivara 07, with an average of 79 copies in the nine rice strains (Fig. 2b).

Copy numbers of mPing (a) and TRIM (b) in the nine rice strains as measured by qPCR (see details in “Materials and methods”)
Insertional polymorphism of TRIM and MITE elements in rice strains
To get more insights into the activity of both TE families, the publicly available sequence of japonica facilitated the design of primers that flanked the 79 TRIMs and 51 mPings, which were used in site-specific PCR (ssPCR) to assess the insertional polymorphism of TRIM and mPing in the nine rice strains. In all cases, the primers for PCR analysis were designed to amplify the TRIM or mPing insertion and flanking host sequences (~100 bp from each side of the intact element). Thus, the expected size of a PCR product will be the size of the TRIM or the mPing insertion plus the flanking sequences. We termed such products as “full site”. In the case of an “empty site”, a lack of a TRIM or mPing insertion, the size of the PCR product will be shorter, containing the flanking sequences alone. An example of a site-specific PCR for TRIM and mPing is shown in Fig. 3. To this end, we have successfully observed clear ssPCR products for 46 of the 51 mPing insertions, and for 55 of the 79 TRIM insertions (Supplemental Table 1).

Site-specific PCR analysis using primers that flanked: a TRIM (AP004811 in Supplemental Table 1) insertion (upper panel), and a mPing (BX000500 in Supplemental Table 1) insertion (lower panel), in nine rice strains: 1 Aikoku Ibaragi 2, 2 Mubo Aikoku, 3 Ginbozu, 4 Gimbozu, 5 Nipponbare, 6 Indica16, 7 O-68-07, 8 Nivara 07, and 9 O. officinalis. NC notes a negative control (H2O was used as a template in PCR). M denotes the 100 bp DNA ladder (Fermentas) that was used. Bands corresponding to either full site or empty site are indicated. Note that bands were isolated from the gel and sequenced for validation
None of the 46 mPing insertions showed a monomorphic insertion (100% polymorphism) in all nine strains, while 32 of the 55 TRIM insertions were monomorphic (41.8% polymorphism). In addition, almost all TRIM insertions were identical in japonica and indica, indicating that TRIMs were inactive after the divergence of the two subspecies, ~0.4 million years ago (Zhu and Ge 2005). In addition, the high similarity between the strains, including those separated geographically and therefore evolutionarily, including the O. officinalis species (genome CC that was separated ~5 million years ago from O. sativa (Zou et al. 2008)), indicates that most probably TRIMs showed little activity throughout rice evolution. Interestingly, the phylogenetic tree that was produced among the nine strains, based on the TRIM markers, significantly classified the Japanese strains in one group (Fig. 4). In contrast, the very high level of polymorphism in the mPing insertions attests to high activity of the element throughout rice evolution as well as in modern rice strains.

Bootstrapped phylogenetic tree of nine rice strains based on the ssPCR results of the 55 TRIM insertions (supplemental Table 1). The level of genetic similarity is indicated at bottom. Bootstrap values are indicated in the branches (p ≤ 0.05 is statistically significant)
Methylation status of TRIM and MITE elements in rice strains
In order to get more insight into the epigenetic regulation of both TRIM and mPing families, we assessed the methylation status of CCGG sites flanking the elements of the two families using TMD (Kashkush and Khasdan 2007; Kraitshtein et al. 2010; Yaakov and Kashkush 2011). Tables 1 and 2 summarize the methylation status of TRIM and mPing elements in the 9 rice strains, respectively. In summary, we found that 52% of the mPing insertions in Nipponbare contain methylated CCGG sites in the flanking sequences, 54% in Aikoku Ibaragi 2, 43% in Mubo Aikoku, 81.5% in Ginbozu, 51% in Gimbozu, 80% in Indica16, 61% in O-68-07, 58% in Nivara 07, and 61% in officinalis. As for TRIM, 72% of the elements in Nipponbare contain methylated CCGG sites in the flanking sequences, 70% in Aikoku Ibaragi 2, 70% in Mubo Aikoku, 70% in Ginbozu, 65% in Gimbozu, 65% in Indica16, 72% in O-68-07, 67% in Nivara 07, and 68% in officinalis.
The high levels of methylation (compared to ~16% of methylation in other rice genomic sequences (Xiong et al. 1999)) as seen by TMD for both TRIM and mPing families indicate that both families are under a strong epigenetic regulation. Also, it can be seen clearly that while TRIMs showed similar levels of methylated CCGG sites in flanking sequences in the nine rice strains (an average of ~69%), there was a significant difference in the methylation levels of mPing (ranging between 43 and 81.5%). The huge CNV and variation in methylation levels of mPing in the various rice strains might indicate that proliferation of mPing might be strongly correlated with its methylation status, as was shown for mPing activity in tissue culture (Ngezahayo et al. 2009), where tissue culture induced the activity of mPing through demethylation (release of methylation). We noticed a significant (p value = 0.035) negative correlation between the copy number of mPing and its methylation levels, with high copy-number rice strains showing lower methylation levels. Alternatively, the massive change in methylation levels between rice strains with high copy number of mPing versus strains with low copy number, could be explained by that strains with high mPing copy number might contain the majority of insertions in euchromatic regions, while strains with low mPing copy number might contain the majority of insertions in heterochromatic regions where the elements mobilization is hindered by the silenced chromatin. Furthermore, the correlation between copy number of mPing and its methylation status can be better tested in additional Gimbozu strains containing highly active mPing elements (over 1,000 copies) (Naito et al. 2006), where we expect to see even lower methylation levels.
In summary, this study shows that while the tiniest class II (mPing) family is the most active TE in rice, the tiniest class I (TRIM) seems to be one of the least active elements in rice. In addition, we observed a strong negative correlation between a TE copy number and its methylation level, which provides additional evidence for the epigenetic regulation of TEs by the host.
References
Clarke KR (1993) Nonparametric multivariate analyses of changes in community structure. Aust J Ecol 18(1):117–143
Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368
Fukao T, Bailey-Serres J, Yeung E (2011) The submergence tolerance regulator SUB1A mediates crosstalk between submergence and drought tolerance in rice. Plant Cell 23(1):412–427
Gehring M, Henikoff S (2008) DNA methylation and demethylation in Arabidopsis. The arabidopsis book, American Society of Plant Biologists, pp 1-14
Han Y, Burnette JM 3rd, Wessler SR (2009) TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res 37(11):e78
Iida S, Morita Y, Choi JD, Park KI, Hoshino A (2004) Genetics and epigenetics in flower pigmentation associated with transposable elements in morning glories. Adv Biophys 38:141–159
Jiang N, Bao ZR, Zhang XY, Hirochika H, Eddy SR, McCouch SR, Wessler SR (2003) An active DNA transposon family in rice. Nature 421(6919):163–167
Jiang N, Feschotte C, Zhang XY, Wessler SR (2004) Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr Opin Plant Biol 7(2):115–119
Kashkush K, Khasdan V (2007) Large-scale survey of cytosine methylation of retrotransposons, and the impact of readout transcription from LTRs on expression of adjacent rice genes. Genetics 177:1975–1985
Kashkush K, Feldman M, Levy AA (2003) Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nature Genet 33(1):102–106
Kikuchi K, Terauchi K, Wada M, Hirano HY (2003) The plant MITE mPing is mobilized in anther culture. Nature 421(6919):167–170
Kishima Y, Takata M, Kiyohara A, Takasu A, Ohtsubo H, Sano Y (2007) Rice transposable elements are characterized by various methylation environments in the genome. BMC Genomics 8:469
Kraitshtein Z, Yaakov B, Khasdan V, Kashkush K (2010) Genetic and epigenetic dynamics of a retrotransposon after allopolyploidization of wheat. Genetics 186(3):801–889
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532
Lockton S, Gaut BS (2009) The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana. J Mol Evol 68(1):80–89
Madlung A, Comai L (2004) The effect of stress on genome regulation and structure. Ann Bot London 94(4):481–495
Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T (2001) Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411(6834):212–214
Naito K, Cho E, Yang GJ, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR (2006) Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci USA 103(47):17620–17625
Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461(7267):1130–1232
Nakazaki T, Okumoto Y, Horibata A, Yamahira S, Teraishi M, Nishida H, Inoue H, Tanisaka T (2003) Mobilization of a transposon in the rice genome. Nature 421(6919):170–172
Ngezahayo F, Xu CM, Wang HY, Jiang LL, Pang JS, Liu B (2009) Tissue culture-induced transpositional activity of mPing is correlated with cytosine methylation in rice. BMC Plant Biol 9:91. doi:10.1186/1471-2229-9-91
Rabinowicz PD, Palmer LE, May BP, Hemann MT, Lowe SW, McCombie WR, Martienssen RA (2003) Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res 13(12):2658–2664
Sabot F, Guyot R, Wicker T, Chantret N, Laubin B, Chalhoub B, Leroy P, Sourdille P, Bernard M (2005) Updating of transposable element annotations from large wheat genomic sequences reveals diverse activities and gene associations. Mol Genet Gen 274(2):119–130
SAS Institute Inc. (1995) JMP statistics and graphic guide
Shan X, Liu Z, Dong Z, Wang Y, Chen Y, Lin X, Long L, Han F, Dong Y, Liu B (2005) Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol Biol Evol 22(4):976–990
Singer T, Yordan C, Martienssen RA (2001) Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene decrease in DNA methylation (DDM1). Genes Dev 15(5):591–602
Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8(4):272–285
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8(12):973–982
Witte CP, Le QH, Bureau T, Kumar A (2001) Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc Natl Acad Sci USA 98(24):13778–13783
Xing JC, Zhang YH, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, Jorde LB (2009) Mobile elements create structural variation: analysis of a complete human genome. Genome Res 19(9):1516–1526
Xiong LZ, Xu CG, Maroof MAS, Zhang QF (1999) Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261(3):439–446
Yaakov B, Kashkush K (2011) Massive alterations of the methylation patterns around DNA transposons in the first four generations of a newly formed wheat allohexaploid. Genome 54(1):42–49
Yang GJ, Nagel DH, Feschotte C, Hancock CN, Wessler SR (2009) Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science 325(5946):1391–1394
Zhu Q, Ge S (2005) Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol 167 (2):632-632 (vol 167, pg 249, 2005)
Zou XH, Zhang FM, Zhang JG, Zang LL, Tang L, Wang J, Sang T, Ge S (2008) Analysis of 142 genes resolves the rapid diversification of the rice genus. Genome Biol 9(3):R49
Acknowledgments
The authors would like to thank Abeer Abu-Siam for help in the lab and Beery Yaakov for helpful discussions. This work was supported by a grant from the Israel Science Foundation (Grant # 142/08) to K. K.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Jordan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Baruch, O., Kashkush, K. Analysis of copy-number variation, insertional polymorphism, and methylation status of the tiniest class I (TRIM) and class II (MITE) transposable element families in various rice strains. Plant Cell Rep 31, 885–893 (2012). https://doi.org/10.1007/s00299-011-1209-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-011-1209-5