
Abstract
The inhibitory activity of bovine pancreatic trypsin inhibitor (aprotinin), a natural polypeptide and a proteinase inhibitor, was demonstrated on gut proteinases of three lepidopteran borers of sugarcane using commercially available aprotinin. A synthetic gene coding for aprotinin, designed and codon optimized for better expression in plant system (Shantaram 1999), was transferred to two sugarcane cultivars namely CoC 92061 and Co 86032 through particle bombardment. Aprotinin gene expression was driven by maize ubiquitin promoter and the plant selection marker used was hygromycin resistance. The integration, expression and functionality of the transgene was confirmed by Southern, Western and insect bioassay, respectively. Southern analysis showed two to four integration sites of the transgene in the transformed plants. Independent transgenic events showed varied levels of transgene expression resulting in different levels (0.16–0.50%) of aprotinin. In in vivo bioassay studies, larvae of top borer Scirpophaga excerptalis Walker (Lepidoptera: Pyralidae) fed on transgenics showed significant reduction in larval weight which indicated impairment of their development. Results of this study show the possibility of deploying aprotinin gene for the development of transgenic sugarcane cultivars resistant to top borer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane, Saccharum spp. hybrid, is an important tropical crop grown for its diverse uses such as production of sugar, ethanol, paper, etc. The cultivated sugarcane is a product of hybridization and repeated clonal selection over a period of time from a large seedling population. This selection process has resulted in clones superior in certain characters like yield and sucrose but deficient in certain other characters like disease or pest resistance. Developing a cultivar with several desirable traits through conventional breeding is a difficult process due to the complexities of the genetic makeup of the crop such as high polyploidy and heterozygosity. A major constraint in increasing productivity of the crop is biotic stresses such as insect pests, especially tissue borers. In India, the shoot borer Chilo infuscatellus Snellen, internode borer Chilo sacchariphagus indicus (K.) (Lepidoptera: Crambidae) and top borer Scirpophaga excerptalis Walker (Lepidoptera: Pyralidae) together pose a major threat to the crop. Among these three borers, top borer infestation causes yield losses of 30–51% (Pandey et al. 1997; Madan and Singh 2001). Even systemic insecticides are generally ineffective against this borer as neonate larvae enter the plants within a few hours of eclosion and remain inside. Also, crop canopy hinders frequent application of insecticides. Since breeding resistant cultivars by conventional methods is a long-drawn process, transgenic technology with insect resistant genes would be a viable alternative.
The advances in genetic transformation technology and knowledge on gene expression have led to rapid progress in using genetic engineering for crop improvement and crop protection against insect pests (Romeis et al. 2006). The potential use of this technology to generate transgenic plants for pest control using different molecules, such as proteinase inhibitors, plant lectins, ribosome inactivating proteins, secondary plant metabolites, delta endotoxins, vegetative insecticidal protein from Bacillus thuringiensis (Bt) and related species, and small RNA viruses, either alone or in combination with the Bt genes (Bates et al. 2005), has now been widely recognized.
Considerable efforts have been made to develop resistance to different borers in sugarcane using genes coding for Cry1Ab (Arencibia et al. 1997), Cry1Ac (Weng et al. 2006), snowdrop lectin (Allsopp and McGhie 1996; Irvine and Mirkov 1997; Nutt et al. 1999) and soybean proteinase inhibitors (Falco et al. 2003). The importance of transgenes as a valuable source of resistance to enhance IPM strategies in sugarcane has been highlighted (Lakshmanan et al. 2005). Bovine pancreatic trypsin inhibitor, also known as aprotinin, a natural polypeptide obtained and purified from cow’s lungs, is widely used as a therapeutic agent in cardiac surgery (Davies et al. 1997). It inhibits serine proteinases (Laskowski and Kato 1980) such as trypsin, chymotrypsin, plasmin and kallikrein (Zhong et al. 1999). Because of the trypsin inhibitory nature (Burgess et al. 2002), it has the potential to be used in genetic transformation to produce insect resistant plants, including sugarcane. In earlier experiments involving aprotinin, a tobacco transgenic expressing about 1.4% of the toxin produced 41% mortality of Spodoptera litura larvae in feeding bioassay (Shantaram 1999). Bovine spleen tryspin inhibitor, a homologue to aprotinin, showed an expression level of 0.5% of total soluble protein in tobacco which affected both survival and growth of late first instar larvae of Helicoverpa armigera (Christellar et al. 2002). Similarly, larvae of Wiseana sp. caterpillar feeding on white cloves expressing aprotinin showed reduced growth rate (Voisey et al. 2001). In this article, we report the first ever work on the development of transgenic sugarcane using aprotinin and its evaluation against sugarcane top borer S. excerptalis. As a prelude to this study, we convinced ourselves of the potential of aprotinin as a candidate for plant protection by examining its effect on three major borers of sugarcane in in vitro bioassays.
Materials and methods
In vitro insect bioassay
Preparation of gut homogenate
Neonate and third instar larvae of C. infuscatellus, C. sacchariphagus indicus and S. excerptalis were used for the assay. In the case of neonate larvae, whole larvae were used as such whereas third instar larvae were etherized, alimentary canals were dissected out and fat bodies adhering to them were carefully teased and removed. The whole larvae or the midguts were homogenized in ice-cold 20 mM Tris–HCl buffer (pH 9.0), centrifuged at 9,400g for 20 min at 4°C and the supernatant was used for studying enzyme activity.
Assay of inhibitory activity of aprotinin on gut proteinases
Assays were performed in small volumes in microtitre plates following the method of Oppert et al. (1997). The protein content of the homogenate was estimated by following Bradford (1976). In the case of neonate larvae, 25 μg of protein was incubated with 0.625, 1.25 and 5 μg of aprotinin whereas for third instar larvae 50 μg of protein from borer gut was incubated with a concentration range of 0.125–10.0 μg of aprotinin. The reaction volume was made up to 100 μl with protein assay buffer [0.5 M Tris–HCl (pH 8.2), 0.02 M CaCl2] and incubated at 37°C for 30 min. To this, 100 μl of 0.5 mg/ml stock of BApNA (N-α-benzoyl-dl-arginine p-nitroanilidine), a trypsin specific substrate prepared in the assay buffer was added. The reaction was allowed to proceed for 30 min at 37°C. Hydrolysis of BApNA by proteinases produces an intense yellow color due to the liberation of p-nitroaniline. Inhibition of proteinases by proteinase inhibitor leads to a decrease in the color intensity, which was measured at 410 nm. The reaction was stopped with the addition of 40 μl of 30% acetic acid solution. In both cases, insect protein without the inhibitor served as control and the protein assay buffer as blank. All tests were repeated thrice.
Plant transformation
Gene construct
The plasmid construct (pSB 203) used for the transformation had the synthetic gene coding for aprotinin under the control of maize ubiquitin promoter and hygromycin as the plant selection marker (Fig. 1). The codon for the aprotinin gene used in the present study was optimized for better expression in plant system. The synthetic gene was made in the laboratory by a combination of synthesis of oligonucleotides in our own machine and a PCR to fill in the gaps. Once the gene was synthesized, it was cloned and sequenced to make sure that there were no errors. Several proof reading polymerases were tried to obtain 100% error-free sequence (Shantaram 1999). Plasmid was isolated and purified from the transformed E. coli by alkali lysis method (Sambrook et al. 1989).

The construct map of pSB 203
Plant tissue culture and callus initiation
Embryogenic calli were produced from 6 to 8-month old healthy shoot tips of the sugarcane (Saccharum spp. hybrid) cultivars Co 86032 and CoC 92061 in the modified MS-I (Murashige and Skoog 1962) medium containing 3 mg/l 2,4-D, 10% coconut milk, 100 mg/l myoinositol with 20 g/l sucrose.
Biolistic bombardment and regeneration
Biolistic bombardment of sugarcane calli was performed following Birch (2000). Friable and embryogenic calli of 2–3 mm size were pretreated in MS-O medium (MSI + Sorbitol 50 g/l + Mannitol 50 g/l) 4 h prior to bombardment. Bombardment of the gene construct pSB 203 was carried out using the Bio-Rad PDS 1000/He biolistic system at a pressure of 1,100 psi of helium. The bombarded calli were incubated in the dark at 25°C for 24 h after which they were transferred to MSI 30 selection media (MS with 30 mg/l hygromycin). After 20–25 days, vigorously growing hygromycin resistant calli were transferred to MSI 50 (MS with 50 mg/l concentration of hygromycin). The hygromycin resistant calli that proliferated on MSI 50 selection media were transferred to regeneration media MSIV (MSI + kinetin 1 mg/l + NAA 0.5 mg/l + 50 mg/l hygromycin) and incubated at 25°C with 16 h light and 8 h dark cycle. When green shoots reached 10–12 cm height, they were transferred to rooting medium (White 1943) with 5 mg/l of hygromycin. Rooted plants were transferred to pots containing a mixture of sterilized sand, soil and farmyard manure. The pots were covered with polyethylene bags to maintain humidity. After a period of 15 days, the acclimatized plantlets were transferred to green house.
Molecular analysis for transgene integration
PCR analysis
Putative transgenics along with the untransformed control plants were subjected to PCR analysis. Genomic DNA was isolated following the method described by Doyle and Doyle (1990). PCR was carried out for detecting the aprotinin gene in the first generation putative transgenics (V0) using aprotinin forward (5′-GGAATTCATGAGGCCAGACT-3′) and Nos reverse (5′-CGTCATGCATTACATGTT-3′) primers to amplify the 330 bp fragment.
Southern transfer and hybridization
The Southern transfer and hybridization was carried out in third vegetative generation transgenics (V3) as described by Sambrook et al. (1989). A measure of 50μg of genomic DNA from putative transgenics and untransformed control plants was digested with HindIII enzyme, electrophoresed on 1% agarose gel and transferred onto nylon membrane (Hybond+, Amersham Biosciences). Southern hybridization was carried out with a α32P-dCTP-labelled fragment containing aprotinin and the promoter sequence (2.2 kb) as probe, excised from pSB 203 by digesting with SacI and HindIII enzymes.
Western blot analysis
Total soluble protein extracted using the buffer [Tris (pH 7.0) 0.05 M, β-mercaptoethanol 2%, glycerol 10%] from the third leaves of V3 transgenics and untransformed control plants were used for Western analysis and enzyme assays. After separation in a 15% SDS-PAGE (Laemmli 1970), the proteins were electro-blotted onto PVDF membrane (Amersham Biosciences) and aprotinin was detected using a rabbit polyclonal antibody to aprotinin (Sigma) following standard protocol (Ausubel et al. 1987). The electro transfer of protein was carried out from gel to membrane for 1 h at 100 V with cooling in a blot transfer apparatus with transfer buffer (5.8 g Tris, 2.93 g glycine, 200 ml methanol and the volume made up to 1 l with water, pH 8.4). The membrane was air dried and after a brief wash in methanol it was incubated in TTBS solution (100 mM Tris–Cl (pH 7.5), 150 mM NaCl and 0.1% Tween 20) for 1 h at room temperature with constant agitation on a rocking platform. After adding the primary polyclonal antibody prepared at 1:1,000 dilution in TTBS, the membrane was incubated for 1 h. The membrane was later washed thrice with TTBS, each wash lasting for 10–15 min. Next, secondary antibody (anti-rabbit IgG conjugated to alkaline phosphatase) was added at 1:500 dilution and the membrane was incubated for another hour. The membrane was then washed with TBS (TTBS without Tween 20). Chromogenic detection was carried out using reagents BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium). The membrane was placed in chromogenic visualization buffer freshly prepared by adding 33 μl of NBT stock (100 mg NBT in 2 ml of 70% DMF) to 5 ml alkaline phosphate substrate buffer [100 mM Tris–Cl (pH 9.5), 100 mM NaCl, 5 mM MgCl2] followed by 17 μl of BCIP stock (100 mg BCIP in 2 ml of 100% DMF). When an indigo colour developed, the reaction was stopped by washing the membrane in distilled water. The membrane was air dried and photographed.
Estimation of aprotinin in transgenics
The protocol followed was similar to that described under in vitro insect bioassay above. A measure of 50 μg of total sugarcane leaf protein was added to 5 μg of bovine pancreatic trypsin in a microtiter plate. The reaction volume was made up to 100 μl using the assay buffer [0.5 M Tris–HCl (pH 8.2), 0.02 M CaCl2] and incubated at 37°C for 30 min. The absorbance was measured at 410 nm in an ELISA reader. The amount of aprotinin was estimated by comparing with the standard values obtained by incubating 5 μg of trypsin with different quantities of aprotinin prepared in the same buffer as used for the extraction of leaf protein. The estimated aprotinin values were expressed as percentage over the total soluble proteins in the sample.
In vivo insect bioassay
Sugarcane leaf spindles having grownup larvae or pupae of top borer were collected from farmers’ fields and maintained on moist sand beds in polyvinyl cages for the emergence of moths. Fresh uninfested cane tops with leaves trimmed were maintained at the center of the cage for oviposition by the moths emerging from infested spindles. Upon oviposition, leaf bits bearing egg masses were separated and maintained on moist filter paper in plastic containers. On eclosion, active neonate larvae were selected and inoculated on 10 transgenic events of CoC 92061, three transgenic events of Co 86032 and untransformed control plants of the respective cultivars. Tops of six canes in each transgenic and untransformed controls were inoculated with three neonate larvae each plant between the +1 and +2 leaves in the crown with a moist camel hairbrush trimmed to a few bristles. For each inoculation, equal numbers of transgenic and untransformed control plants were taken in accordance with the number of neonate larvae obtained on that day of eclosion. Prior to the inoculation of larvae, the plants were checked and freed of general predators. Shoot tops of test plants were excised on the 20th day of inoculation of neonate larvae and parameters, such as the number of midribs tunneled, length of the tunnel, length of unfed spindle core and weight of larvae in the inner core were recorded in transgenics of Co 86032 and CoC 92061 along with the respective untransformed control plants.
Data analysis
Data were analyzed following Gomez and Gomez (1984). Top borer feeding parameters were subjected to square root transformation, analysis of variance and Duncan’s multiple range test for mean comparison. Further, aprotinin content was correlated with feeding parameters to assess their interdependence.
Results
Inhibitory activity of aprotinin on insect gut trypsin
Inhibitory activity of aprotinin was represented as percentage of residual trypsin obtained after the hydrolysis of BapNA by trypsin in in vitro bioassays. At the lowest aprotinin concentration of 0.625 μg, the residual trypsin obtained in neonate larvae of shoot borer, internode borer and top borer was 33.6, 24.8 and 9.3%, respectively (Fig. 2). The residual trypsin for third instar of shoot borer, internode borer and top borer at the lowest aprotinin concentration of 0.125 μg was 55.0, 65.0 and 20.7% respectively. The amount of aprotinin needed to bring about 50% inhibition of gut proteinases of third instar larvae was 0.25 μg for shoot borer and 2.5 μg for internode borer whereas 0.125 μg was sufficient to bring about nearly 80% inhibition in top borer (Fig. 3). Thus, in both stages of the three different borers, aprotinin showed the highest inhibitory effect on gut proteinases of top borer.

Inhibition of trypsin by different concentrations of aprotinin in gut bioassay of neonate larvae of sugarcane borers

Inhibition of trypsin by different concentrations of aprotinin in gut bioassay of third instar larvae of sugarcane borers
Sugarcane transformation
Transgenics expressing aprotinin were generated for two sugarcane cultivars, i.e. Co 92061 and Co 86032 through particle bombardment. In both cultivars, around 10% transformation frequency (number of transgenic events over the number of calli bombard) was obtained. Twenty-three putative transgenics that showed resistance to hygromycin were established in green house. Out of these, 16 proved to be true transgenics expressing aprotinin.
PCR and Western analysis
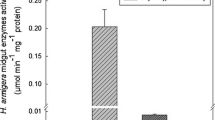
Sixteen PCR positive plants amplifying the expected fragment of 330 bp (Fig. 4) were subjected to Western analysis to examine the transgene expression and integrity of aprotinin produced by the transgenic plants (Fig. 5). This experiment showed the presence of a 6.5 kDa protein in the transgenics with a clear band similar to that of the commercial aprotinin used as a positive control, which was absent in the untransformed control plant.

PCR analysis of transgenics. Lanes 1–3 T1–T3 (cv. CoC 92061), 4–6 P1–P3 (cv. Co 86032), 7 untransformed control plant (cv. Co 86032), 8 positive control (pSB203), 9 1 kb DNA marker (Fermentas)

Western blotting analysis for the expression of aprotinin in transgenics. Lanes 1 positive control (commercial aprotinin), 2 untransformed control (cv. Co 86032), 3–4 T1–T2 (cv. CoC 92061), 5–6 P1–P2 (cv. Co 86032)
Southern analysis
In Southern analysis with the genomic DNA of three transgenic events of CoC 92061 and three of Co 86032 expressing higher levels of aprotinin, all the transgenic lines defined two or more integration sites of the transgene with a maximum of four (Fig. 6).

Southern blot analysis showing stable integration of 2.2 kb fragment containing aprotinin with the ubi promoter in transgenics of cv. Co 86032 and CoC 92061. DNA samples were digested with HindIII. Lanes 1 untransformed control, 2–4 transgenics of cv. Co 86032, 5 uncut transgenic of cv. Co 86032, 6 positive control, 7 untransformed control, 8–10 transgenics of cv. CoC 92061
Quantification of aprotinin expression in transgenics
The amount of aprotinin ranged from 0.16 to 0.50% in transgenic plants of CoC 92061 (Table 1) and from 0.20 to 0.26% in transgenic plants of Co 86032 (Table 2).
In vivo insect bioassay
In CoC 92061, the transgenics T3 and T7 had significantly lower number of midribs tunneled than T1; the transgenics T3, T5 and T6 had significantly shorter tunnel length than T9 (Table 1). In Co 86032, however, the number of tunnels and tunnel length did not differ significantly between transgenic and untransformed control plants (Table 2). The unfed spindle core measured as the distance at which larvae were located from the meristem after 20 days of feeding, an index of the progress in larval feeding activity in the core of the meristem, did not vary between transgenic and untransformed control plants of both CoC 92061 and Co 86032. Neonate larval mortality was seen in the midribs of transgenic plant P3 of Co 86032 only (Fig. 7). Both CoC 92061 and Co 86032 showed significant reduction in mean larval weight compared to control (Fig. 8). However, in CoC 92061, larval weight in different transgenics varied widely with overlapping significant differences while in Co 86032 transgenics did not show significant differences. Aprotinin content, mean tunnel length, unfed spindle core and mean larval weight did not show congruence among different transgenics in either cultivar. For example, larval weight in the transgenics T1 and T4 was significantly less than that in T2 and control of CoC 92061, though the number and length of tunnels were on par or even higher than in control plants. Correlation analysis among these variables showed significant negative relationship between aprotinin content and larval weight in CoC 92061 (Fig. 9). However, aprotinin content was not significantly related to any other parameter.

Dead top borer larva in the midrib of transgenic P3 (cv. Co86032)

Top borer larvae collected from a untransformed control and b transgenic (T4) sugarcane (cv. CoC 92061) 20 days after infestation

Correlation between aprotinin content and top borer larval weight in cv. CoC 92061
Discussion
The objective of the present study was to generate transgenic sugarcane expressing aprotinin that would show resistance to borer pests. As a prelude, inhibitory activity of aprotinin was evaluated against three sugarcane borers in in vitro bioassay as a simple approach to evaluate the potential effectiveness of proteinase inhibitors (Christellar and Shaw 1989) though in vivo methods have also been used for bioassay (Baszczynski et al. 1998). Although the insecticidal action of proteinase inhibitors is largely attributed to inhibition of digestive enzymes in the insect gut, depletion of essential amino acids due to over-secretion of digestive enzymes in the presence of inhibitors is suggested to cause most of the toxicity symptoms (Ryan 1990). Regardless of the mechanism of toxicity, aprotinin was more effective on gut proteinases of top borer than shoot borer and internode borer. Such differential effectiveness of aprotinin suggested the qualitative and quantitative differences in proteinases characteristic of their taxonomic affiliation, the genus Scirpophaga being more susceptible than the genus Chilo. In a similar in vitro bioassay with insects belonging to different taxonomic groups, including a species of Scirpophaga, aprotinin inhibited midgut proteinases to varying levels (Shantaram 1999) reaffirming the differential response of different species of insects. Our results (Figs. 2, 3) showed that trypsin inhibition in both neonate and third instar larvae of top borer was not dependent on aprotinin concentration, though the other two borers displayed some dosage-dependent response. However, at a comparable concentration of 1.25 μg of aprotinin, the difference in trypsin inhibition between neonate larvae and third instar larvae of top borer was only about 10%. This indicated that transgenics expressing aprotinin can be effective against neonate as well as grownup stages of top borer.
Encouraged by the positive results of in vitro assays of aprotinin, two popular cultivars of sugarcane were transformed with the gene coding for it after codon optimizing for better expression in plants. Southern hybridization in six transgenics showed multiple integration of the transgene as evidenced by multiple bands. Direct transformation methods are known to produce complex events in which multiple copies of the introduced DNA get integrated at one or several loci in the recipient genome (Christou 1992; Saul and Potrykus 1990) and transgene copy number is known to influence gene expression either positively or negatively (Hobbs et al. 1993). In Southern analysis of maize transgenics expressing aprotinin, only one transgenic line had five or fewer copies of the transgene whereas 20 or more copies of aprotinin and bar genes were co-integrated in others (Zhong et al. 1999). In the present study, Southern analysis showed that the integration sites varied between two and four which is below the expectation of above five in case of transgenics produced through particle bombardment. The fewer integration sites observed could possibly be a result of stringent selection in the early stages in which transgenics with higher integration sites/copy numbers, and the consequent lack of expression of the selectable marker due to gene silencing, would have been eliminated. Southern analysis performed for a limited number of transgenics in the present study precluded the establishment of a relationship between expression of aprotinin and the copy number. Western blot analysis has demonstrated that the size of aprotinin expressed in transgenics was same as that of the commercial aprotinin, which is of mammalian origin, suggesting their structural similarity. Aprotinin was detected in varying levels (0.16–0.50%) in all transgenics that expressed the transgene. Such variation in the expression of different proteinase inhibitors was observed in transgenics of other crops too: aprotinin (0.012–0.44%) in seeds of maize (Zhong et al. 1999); potato proteinase inhibitor (0.35–2.3%) (Duan et al. 1996), cowpea trypsin inhibitor (CpTi) (0.3–2.7%) (Xu et al. 1996), corn cystatin (2.4%) (Irie et al. 1996) and aprotinin (0.4–1.3%) (Shantaram 1999) in rice; CpTi (up to 1%) (Hilder et al. 1987) and aprotinin (0.3–1.9%) (Shantaram 1999) in tobacco. The expression levels of aprotinin in the present study were comparable to those in maize reported by Zhong et al. (1999) whereas they were lower than those in rice and tobacco (Shantaram 1999).
In the in vivo bioassay with neonate larvae of top borer on transgenics expressing varying levels of aprotinin, larval feeding parameters did not show definite trends, except for a few significant differences among transgenics of CoC 92061 for the number of midribs tunneled and tunnel length. Although the number or length of tunnels did not differ between resistant and susceptible groups of plants (Mukunthan 1984), a later study on the mechanism of resistance to the borer established that resistance operated only in the midrib and not in the spindle (Mukunthan 1990). Despite the exceptions of a few transgenics in the present study, midrib did not seem to be the site of active resistance as there was no evidence of mortality of neonate larvae in midribs of these plants. Also, the lack of variation in unfed spindle core did not indicate spindle too as the active site of resistance. This suggested the lack of differential expression of aprotinin in midrib and spindle, probably due to the constitutive promoter used, to levels that could inhibit larval growth in the two successive feeding stages. The significant larval weight loss in transgenic plants of both Co 86032 and CoC 92061 indicated that aprotinin had a cumulative antibiotic effect on larval growth as a consequence of its feeding in midrib and spindle. Maximum larval weight reduction was greater in CoC 92061 (99.8%) than in Co 86032 (82.9%), probably due to the inherent varietal difference in borer resistance, besides the interactive effect of aprotinin integration. Although larval developmental rate did not differ between resistant and susceptible genotypes in top borer (Mukunthan and Mohanasundaram 1996), distinct poor weight gain was observed in internode borer larvae on traditional resistant cultivars which was attributed to antibiosis as the mechanism of resistance (David 1979). Leaf tissues from sugarcane transgenics with soybean kunitz trypsin inhibitor and soybean Bowman–Birk inhibitor significantly retarded the growth of Diatraea saccharalis larvae as compared to leaf tissue from untransformed plants (Falco et al. 2003). In other crops too, proteinase inhibitors generally affected survival and growth of lepidopteran larvae (Voisey et al. 2001; Christeller et al. 2002), though mortality was also reported (Shantaram 1999) apparently due to higher protein expression levels. These observations indicate that proteinase inhibitors, in general, affect the growth and development of larvae, though not result in their mortality. This contention also gains support from the significant reduction in larval weight gain in the taxonomically different cane grub Antitrogus consanguineus fed on roots of transgenic sugarcane expressing potato proteinase inhibitor II or snowdrop lectin (Nutt et al. 1999; Allsopp et al. 2000). Transgenic plants rarely result in 100% control but help to retard insect growth and development (Estruch et al. 1997) thereby reducing the loss inflicted by them on crop plants. The significant reduction in larval weight (up to 99.8%) observed in the present study could confer two advantages: reduced intensity of damage to the crop in the current brood and decreased populations in the subsequent broods. This would be of greater advantage in subtropical India where top borer exhibits distinct brood pattern.
The present studies indicate that introduction and expression of aprotinin encoding gene into sugarcane cultivars can be an effective strategy for conferring considerable level of protection against top borer. Despite the lower susceptibility of shoot borer and internode borer to aprotinin in in vitro studies, it is possible that transgenics expressing the toxin may show a lower level of field tolerance to these borers too. Engineering aprotinin or other proteinase inhibitors in conjunction with Bt toxins or lectins, by either cross breeding of primary transformants or multiple gene insertions, would probably enhance the resistance levels of sugarcane transgenics. This approach would also address the problem of resistance to proteinase inhibitors in insects (Jongsma and Bolter 1997).
References
Allsopp PG, McGhie TK (1996) Snowdrop lectin and wheatgerm lectins as antimetabolites for the control of sugarcane whitegrubs. Entomol Exp Appl 80:409–414
Allsopp PG, Nutt KA, Geijskes RJ, Smith GR (2000) Transgenic sugarcane with increased resistance to cane grubs. In: Allsopp PG, Suasa-ard W (eds) Proceedings of the International Soceity of Sugar Cane Technologists, Sugar Cane Entomology Workshop. Khon Kaen, Thailand, pp 63–67
Arencibia A, Vazquez RI, Prieto D, Tellez P, Carmona ER, Coego A, Hernandez L, DelaRiva GA, SelmanHousein G (1997) Transgenic sugarcane plants resistant to stem borer attack. Mol Breed 3:247–255
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1987) Current protocols in molecular biology. Green Publishing Associates/Wiley Interscience, New York
Baszczynski C, Czapla T, Hood E, Meyer TE, Peterson D, Rao AG, Register JC III, Witcher D, Howard J (1998) Commercial production of aprotinin in plants. Prodigene Inc., US patent 5,824,870, 20 October 1998
Bates SL, Zhao JZ, Roush RT, Shelton AM (2005) Insect resistance management in GM crops: past, present and future. Nat Biotechnol 3:57–62
Birch RG (2000) Sugarcane transformation: microprjectile bombardment of embryogenic callus and geneticin selection. Birch laboratory protocol, pp 1–6
Bradford MM (1976) A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254
Burgess EPJ, Lovei GL, Malone LA, Nielsen JW, Gatehouse HS, Christellar JJ (2002) Pray mediated effects of the protease inhibitor aprotinin on the predatory carabid beetle Nebria brevicollis. J Insect Physiol 48:1093–1101
Christellar JT, Shaw BD (1989) The interaction of a range of serine proteinase inhibitors with bovine trypsin and Costelytra zealandica trypsin. Insect Biochem 19:233–241
Christellar JT, Burgess EPJ, Mett V, Gatehouse HS, Markwick NP, Murray C, Malone LA, Wright M, Philip BA, Watt D, Gatehouse LN, Lovei GL, Shannon AL, Phung MM, Watson L, Liang WA (2002) The expression of mammalian proteinase inhibitor, bovine spleen trypsin inhibitor (SI) in tobacco and effects on Helicoverpa armigera larvae. Transgenic Res 11:161–173
Christou P (1992) Genetic transformation of crop plants using microprojectile bombardment. Plant J 2:275–281
David H (1979) A critical evaluation of the factors associated with resistance to internode borer, Chilo sacchariphagus indicus (Kapur) in Saccharum sp. and allied genera and hybrid sugarcane. PhD thesis. Calicut University, Calicut
Davies MJ, Allen A, Kort H, Weerasena NA, Rocco D, Paul CL, Hunt BL, Elliott MJ (1997) Prospective, randomized, double-blind study of high-dose aprotinin in pediatric cardiac operations. Ann Thorac Surg 63:497–503
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Duan X, Li X, Xue Q, Abo el Saad M, Xu D, Wu R (1996) Transgenic rice plants harbouring an introduced potato protease inhibitor II gene are insect resistant. Bat Biotechnol 14:494–498
Estruch JJ, Carozzi NB, Desai N, Duck NB, Warren GW, Koziel MG (1997) Transgenic plants an emerging approach to pest control. Nat Biotechnol 15:137–141
Falco MC, Silva-Filho MC (2003) Expression of soybean proteinase inhibitors in transgenic sugarcane plants: effects on natural defense against Diatraea saccharalis. Plant Physiol Biochem 41:761–766
Gomez KA, Gomez AA (1984) Statistical procedures for agricultural Research, 2nd edn. John Wiley and Sons, New York, p 680
Hilder VA, Gatehouse AMR, Sheerman SE, Baker RT, Boulter D (1987) A novel mechanism of insect resistance engineered into tobacco. Nature 330:160–163
Hobbs SLA, Warkentin TD, de Long CMO (1993) Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol Biol 21:17–26
Irie K, Hosoyama H, Takeuchi T, Iwabuchi K, Watanabe H, Abe M, Abe K, Arai S (1996) Transgenic rice established to express corn cystatin exhibits strong inhibitory activity against insect gut proteinases. Plant Mol Biol 30:149–157
Irvine JE, Mirkov TE (1997) The development of genetic transformation of sugarcane in Texas. Sugar J 60:25–29
Jongsma MA, Bolter C (1997) The adaptation of insects to plant protease inhibitors. J Insect Physiol 43:885–895
Lakshmanan P, Geijskes RJ, Aitken KS, Grof CLP, Bonnett GD, Smith GR (2005) Sugarcane biotechnology: the challenges and opportunities. In Vitro Cell Dev Biol Plant 41:345–363
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage: T4. Nature 227:680–685
Laskowski M, Kato I (1980) Protein inhibitors of proteinases. Annu Rev Biochem 49:593–626
Madan YP, Singh M (2001) Assessment of extent of damage and losses caused by top borer (Scirpophaga excerptalis Walker) in sugarcane in Haryana. Indian Sugar 51(2):99–102
Mukunthan N (1984) Tensile strength of midribs in relation to the top borer resistance. Proc Annu Conf Deccan Sugar Technol Assoc 34:109–116
Mukunthan N (1990) Resistance studies in sugarcane to top borer Scirpophaga excerptalis Walker. PhD Thesis. Tamil Nadu Agricultural University, Coimbatore
Mukunthan N, Mohanasundaram N (1996) The development of top borer in different host plants. Sugarcane 6:11–13
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Nutt KA, Allsopp PG, McGhie TK, Shepherd KM, Joyce PA, Taylor GO, McQualter RB, Smith GR, Hogarth DM (1999) Transgenic sugarcane with increased resistance to canegrubs. Proc Aust Soc Sugar Cane Technol 21:171–176
Oppert B, Krammer KJ, McGaughey WH (1997) Rapid microplate assay for substrates and inhibitors of proteinase mixtures. Biotechniques 23:70–72
Pandey KP, Sharma BL, Singh RG (1997) Effect of different density of eggmass of top borer (Scirpophaga excerptalis Walker) on growth, yield and quality of sugarcane. Entomology 22:247–249
Romeis J, Meissel M, Bigler F (2006) Transgenic crops expressing Bacillus thuringiensis toxin and biological control. Nat Biotechnol 4:63–71
Ryan CA (1990) Protease inhibitors in plants: genes for improving defenses against insects and pathogens. Annu Rev Phytopathol 28:425–449
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Saul MW, Potrykus Y (1990) Direct gene transfer to protoplasts: fate of the transferred genes. Dev Genet 11:176–181
Shantaram B (1999) Engineering of bovine pancreatic trypsin inhibitor gene for plant expression. PhD Thesis. University of Madras, Chennai, India
Voisey CR, Dudas B, Biggs R, Burgess EPJ, Wigley PJ, McGregor PG, Lough TJ, Beck DL, Forster RLS, White DWR (2001) Transgenic pest and disease resistant white clover plants. In: Spangenberg G (ed) Molecular breeding of forage crops. Kluwer Academic Publishers, London, pp 239–250
Weng LX, Deng H, Xu JL, Li Q, Wang LH, Jiang Z, Zhang HB, Li Q, Zhang LH (2006) Regeneration of sugarcane elite breeding lines and engineering of stem borer resistance. Pest Manag Sci 62:178–187
White PR (1943) Handbook of plant tissue culture. The Ronald Press Co., New York
Xu DP, Xue QZ, McElroy D, Mawal Y, Hilder VA, Wu R (1996) Constitutive expression of a cowpea trypsin ihibitor gene, CpTi in transgenic rice plants confers resistance to two major rice insect pests. Mol Breed 2:167–173
Zhong GY, Peterson D, Delaney DE, Bailey M, Witcher DR, Register JCIII, Bond D, Li CP, Marshall L, Kulisek E, Ritland D, Meyer T, Hood EE, Howard JA (1999) Commercial production of aprotinin in transgenic maize. Mol Breed 5:345–356
Acknowledgments
The authors wish to thank Dr. M. Karunakaran for the help rendered in Southern analysis and Mrs. R. Nirmala for assistance in bioassays.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Lakshmanan.
Rights and permissions
About this article
Cite this article
Christy, L.A., Arvinth, S., Saravanakumar, M. et al. Engineering sugarcane cultivars with bovine pancreatic trypsin inhibitor (aprotinin) gene for protection against top borer (Scirpophaga excerptalis Walker). Plant Cell Rep 28, 175–184 (2009). https://doi.org/10.1007/s00299-008-0628-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-008-0628-4