
Abstract
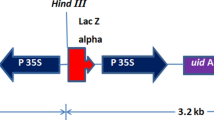
A reliable genetic transformation protocol via somatic embryogenesis has been developed for the production of fertile, herbicide-resistant opium poppy plants. Transformation was mediated by Agrobacterium tumefaciens using the pCAMBIA3301 vector, which harbors the phosphinothricin acetyltransferase (pat) gene driven by a tandem repeat of the cauliflower mosaic virus (CaMV) 35S promoter and the β-glucuronidase (gus) structural gene driven by a single copy of the CaMV 35S promoter between left- and right-border sequences. Co-cultivation of explants and A. tumefaciens was performed in the presence of 50 μM ATP and 50 μM MgCl2. Root explants pre-cultured on callus induction medium were used for transformation. Herbicide-resistant, proliferating callus was obtained from explants on a medium containing both 2,4-dichlorophenoxyacetic acid (2,4-D) and 6-benzyladenine (BA). Globular embryogenic callus, induced by removal of the BA from the medium, was placed on a hormone-free medium to form somatic embryos, which were converted to plantlets under specific culture conditions. Plantlets with roots were transferred to soil, allowed to mature and set seed. Both pat and gus gene transcripts, and PAT and GUS enzyme activities were detected in the transgenic lines tested. Histochemical localization of GUS activity in T1 opium poppy plants revealed transgene expression in most tissues of all plant organs. The protocol required 8–12 months to establish transgenic T1 seed stocks and was developed using a commercial opium poppy cultivar that produces high levels of pharmaceutical alkaloids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Opium poppy (Papaver somniferum L.) is one of humankind’s oldest medicinal plants and remains the only source of the narcotic analgesics and antitussive drugs morphine and codeine, and a number of other benzylisoquinoline alkaloids of pharmaceutical significance, such as the vasodilator and smooth muscle relaxant papaverine, and the antitussive and antitumorogenic agent noscapine. In addition, thebaine is used extensively as a starting material for the production of oxycodone and other semi-synthetic analgesic opiates. Australia, India, France, Turkey, Spain, Hungary and the Czech Republic are the major licit producers of opium and poppy straw for the extraction of pharmaceutical alkaloids. In general, poppy straw is harvested in temperate zones, whereas opium is collected in more tropical locations. Several other European countries cultivate opium poppy for the production of confectionary seeds (Hagel et al. 2007).
The early domestication and aggressive breeding of opium poppy have guided the establishment of modern phenotypes. Classic breeding programs have produced large germplasm collections, although limited genetic variation with respect to chemical and agronomic traits has been reported for Indian (Singh and Khanna 1991) and European (Dubedout 1993) cultivars due to the relatively narrow base of opium poppy genotypes (Levy and Milo 1997). Key requirements for the commercial cultivation of opium poppy are a high alkaloid percentage in the straw, and a high alkaloid yield per hectare. Alkaloid production per hectare is important because increases in production depend on additional yield, rather than increased cultivation area, and the return per hectare must be competitive compared with other crops. The major factors affecting the yield of poppy straw are genotype, nitrogen and phosphorus nutrition, soil water status, weeds, pest and disease. Future prospects for weed control include the use of transgenic opium poppy rendered resistant to herbicides (Chitty et al. 2003).
Despite difficulties in the regeneration of intact plants from cultured tissues, the genetic transformation of opium poppy organs and plants has been reported. Park and Facchini (2000a) used Agrobacterium rhizogenes to produce transgenic hairy roots from wounded opium poppy seedlings. Similarly, Le Flem-Bonhomme et al. (2004) used A. rhizogenes to infect wounded hypocotyls and induce hairy root formation. Although Park and Facchini (2000a) showed that agar-solidified media could be used to induce transgenic roots, Le Flem-Bonhomme et al. (2004) suggested that opium poppy hairy roots could be obtained only in liquid medium without growth regulators. Nessler (1998) described the regeneration of transgenic opium poppy plants using either Agrobacterium tumefaciens or microparticle bombardment, although the reported work was preliminary. Park and Facchini (2000b) described the A. tumefaciens-mediated transformation of hypocotyls and the regeneration of opium poppy plants by shoot organogenesis, but the efficiency of the protocol was low. Chitty et al. (2003) reported the infection of hypocotyls with A. tumefaciens to produce embryonic callus, which could be used to regenerate transgenic opium poppy plants through somatic embryogenesis. Although the technique was reported to be efficient and effective across a range of opium poppy cultivars, the only innovations in terms of protocol optimization involved the inclusion of a pH buffer in the culture medium and bottom cooling of the culture vessels.
The regeneration of opium poppy plants through somatic embryogenesis has been reported with variations in hormone concentration and explant source (Nessler 1982; Wakhlu and Bajwa 1986; Dieu and Dunwell 1988). However, the conversion of opium poppy somatic embryos usually resulted in plantlets lacking roots (Ovecka et al. 1997; Kassemi and Jacquin 2001). Moreover, the production of secondary embryos or adventitious shoots during the somatic embryogenesis of opium poppy on hormone-free media has also been reported (Ovecka et al. 1997, 2000). Similar regeneration effects were reported for other members of the Papaveraceae, including Papaver bracteatum (Ilahi and Ghauri 1994), P. orientale (Schuchmann and Wellmann 1983), Corydalis yanhusuo (Sagare et al. 2000) and Eschscholzia californica (Park and Facchini 2000c, 2001). The prevention of such phenomena is necessary to establish an efficient transformation system for opium poppy with potential genetic engineering and genomics applications.
Reliable genetic transformation methods are a key component in the application of biotechnology to the opium poppy industry. Combined with a growing repository of cloned genes and an expanding knowledge of the regulation of alkaloid biosynthetic pathways, metabolic engineering via genetic transformation could provide unprecedented capacity to engineer new plant varieties with commercially desirable traits and expand the frontiers of the licit opium poppy industry. In this paper, we describe the establishment of a genetic transformation protocol based on the A. tumefaciens-mediated infection of pre-cultured explants and the regeneration of transgenic, herbicide-resistant opium poppy plants.
Materials and methods
Preparation of Agrobacterium tumefaciens
The pCAMBIA3301 transformation vector, which harbors the phosphinothricin acetyltransferase (pat) gene driven by a tandem repeat of the cauliflower mosaic virus (CaMV) 35S promoter and the β-glucuronidase (gus) gene driven by a single copy of the CaMV 35S promoter between left- and right-border sequences, was mobilized by electroporation in A. tumefaciens strain EHA105. Bacterial cultures were grown to mid-log phase (A 600 = 0.5) on a gyratory shaker at 180 rpm, in liquid Luria-Bertani medium (1% [w v−1] trypotone, 0.5% [w v−1] yeast extract, 1% [w v−1] NaCl, pH 7.0) containing 50 mg L−1 kanamycin. Cells are collected by centrifugation at 3,000 rpm for 10 min, washed in inoculation medium containing B5 salts and vitamins (Gamborg et al. 1968), 1% (w v−1) glucose, 2% (w v−1) sucrose, 50 μM ATP, and 50 μM MgCl2, re-collected by centrifugation for 10 min, and re-suspended at a density of A 600 = 0.25 in fresh inoculation medium.
Seed sterilization and germination
Seeds of opium poppy (Papaver somniferum L.) cultivar Louisiana were sterilized for 1 min in 70% (v v−1) ethanol, and 10 min in 5.25% (v v−1) sodium hypochlorite solution, then rinsed three times in sterilized distilled water. Seeds were plated on solid germination medium containing MS salts and vitamins, 2% (w v−1) sucrose, and 0.8% (w v−1) Difco noble agar (BD Biosciences, Mississauga, Canada). Seeds were germinated at 25°C under standard cool white fluorescent tubes (Sylvania Gro-Lux Wide Spectrum, Mississauga, Canada) with a flux rate of 35 mmol s−1 m−2 and a 16-h photoperiod.
Pre-cultivation and infection of opium poppy callus
Roots from 7-day-old seedlings were pre-cultured in the dark for 3–4 weeks on callus induction medium, which consisted of MS salts (Murashige and Skoog 1962), White’s organic supplements (White 1967), 0.5 g L−1 myo-inositol, 2.5% (w v−1) sucrose, 2.5 μM 2,4-dichlorophenoxyacetic acid (2,4-D), 2.5 μM 6-benzyladenine (BA), and 0.8% (w v−1) Difco noble agar, pH 5.7. Pre-cultured explants were wounded in the presence of A. tumefaciens using a new sterile scalpel blade, and were then co-cultivated for 18 h at 75 rpm on a gyratory shaker in co-cultivation medium consisting of B5 salts and vitamins, 1% (w v−1) glucose, 2% (w v−1) sucrose, 50 μM ATP, 50 μM MgCl2, 2.5 μM 2,4-D, and 2.5 μM BA, pH 5.7. Tissues were then washed with co-cultivation medium containing 300 mg L−1 timentin and blotted on sterilized filter paper to remove excess liquid. Tissues were transferred to callus induction medium containing 300 mg L−1 timentin and maintained in the dark for 7 days to eliminate the A. tumefaciens.
Selection of transgenic callus
Explants were transferred to the medium consisting of MS salts, White’s organic supplements, 0.5 g L−1 myo-inositol, 2.5% (w v−1) sucrose, 2.5 μM 2,4-D, 2.5 μM BA, 10 mg L−1 phosphinothricin (PPT; glufosinate ammonium), 300 mg L−1 timentin, and 0.8% (w v−1) Difco noble agar, pH 5.7. Initial proliferating callus was transferred to fresh selection medium every 4 weeks for 3 months to produce herbicide-resistant callus, from which white globular callus was induced 4 to 6 weeks after transfer to selection medium containing 5 μM 2,4-D, but lacking BA.
Conversion and rooting of somatic embryos
White globular callus was transferred to hormone-free medium consisting of MS salts, 3% (w v−1) sucrose, White’s organic supplements, 0.5 g L−1 myo-inositol, 0.5 g L−1 l-glutamine, and 0.8% (w v−1) Difco noble agar. Cultures were kept in the dark at 24°C. Somatic embryos at the cotyledonary stage were incubated at 15°C under a 16-h photoperiod in 500 μM indolebutyric acid (IBA) for 5 h, then transferred to Magenta boxes containing MS medium supplemented with 2% (w v−1) sucrose and 0.8% (w v−1) Difco noble agar. Sterilized filter paper was placed on the surface of the medium to prevent direct contact between the somatic embryos and the agar.
Real-time PCR
Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). Real-time PCR was performed using an Applied Biosystems (Foster City, CA, USA) 7300 Real-Time PCR System. Reverse transcription was performed at 42°C for 90 min using 2.5 μM random hexamers as primers, 500 μM dNTPs and 50 U of reverse transcriptase (Fermentas, Burlington, Canada), after denaturing the RNA-primer mix at 70°C for 5 min. The primer sequences used were CCCGCAACGCCTACGA (pat5-qRT-f1) and GCCGGTGGGAGACGTACA (pat6-qRT-r1) for the pat gene, CGGAAGCAACGCGTAAACTC (gus-qRT-f1) and GCGTCGCAGAACATTACATTGA (gus-qRT-r1) for the gus gene, and TGCTCCAGTTCTTGACTGTC (el1a-qRT-f1) and GTCTCTACAACCATGGGCTTG (el1a-qRT-r1) for the elongation factor 1a (elf1a) gene. Reactions for real-time PCR using SYBR green detection included 5 μL of SYBR green PCR Master Mix (Applied Biosystems), 300 nM of each forward and reverse primer, and 10 ng of cDNA template in a final reaction volume of 10 μL. All PCR reactions were prepared in triplicate. Forty amplification cycles, consisting of 95°C for 20 s and 60°C for 1 min were performed. The real-time PCR data were plotted as the ΔRn fluorescence signal versus cycle number. An arbitrary threshold was set during the exponential phase of the ΔRn versus the cycle number plot, and the Ct values defined as the cycle number at which the ΔRn crosses this threshold. The ΔCt was calculated using the equation ΔCt = Ct+ − Ct−, where Ct+ is the fluorescence signal of amplicons derived using pat or gus primers and Ct− is the fluorescence signal from primers for the constitutive elf1a control (Auriac and Timmers 2007).
PAT enzyme assay
Tissues were extracted in 50 mM sodium phosphate buffer, pH 7.5, containing 20 mM NaCl and 1 mg L−1 bovine serum albumin. Debris was removed by centrifugation and the supernatant was used to assay PAT activity. Reactions consisted of 37 mM phosphinothricin (PPT; glufosinate ammonium), 0.1 mCi (1.82 nmol) [acetyl-1-14C]-acetyl coenzyme A, 25 μL of protein extract, and extraction buffer resulting in a total volume of 30 μL (Trieu and Harrison 1996). Assays were incubated for 30 min at 37°C and, subsequently, 15 μL of the reaction was applied to a silica gel 60 F254 thin layer chromatography (TLC) plate (EM Science, Gibbstown, NJ, USA), which was developed in 1-propanol:NH4OH:H2O (6:1:3; v v−1 v−1). [14C]-Acetylated PPT produced (R f = 0.40) was quantified using a Bio-Imaging Analyzer (FUJIX BAS 1000, Fuji Film, Tokyo, Japan).
GUS enzyme assay
Tissues were extracted in 50 mM potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, and 10 mM β-mercaptoethanol. Debris was removed by centrifugation and the supernatant was used to assay GUS activity. 4-Methylumbelliferyl-β-D-glucuronide (MUG) was added at a final concentration of 1.3 μM to the GUS fluorometric assay buffer (50 mM NaPO4 buffer, pH 7.0, 10 mM β-mercaptoethanol, 10 mM EDTA, 0.1% [w v−1] sodium lauryl sarcosine, and 0.1% [w v−1] Triton X-100). Assays were performed on 50 μL of protein extract for 3 h at 37°C and stopped with a ten volumes of 0.2 M Na2CO3. The cleavage of 4-methylumbelliferone (MU) from MUG was monitored using a fluorescence spectrophotometer (Hitachi F-2000, Tokyo, Japan). Protein concentrations were determined by the method of Bradford (1976) using BSA as the standard.
GUS histochemical staining
Histochemical staining for GUS activity was performed using the method of Jefferson et al. (1987) modified as recommended by Kosugi et al. (1990). Tissues were fixed in a 0.35% (v v−1) formaldehyde solution containing 10 mM MES, pH 7.5, and 300 mM mannitol for 1 h at 20°C, rinsed three times in 50 mM sodium phosphate, pH 7.5, and subsequently incubated in 50 mM sodium phosphate, pH 7.5, 10 mM EDTA, 300 mM mannitol, pH 7.0, and 1 mM 5-bromo-4-chloro-3-indolyl-β-D-glucuronide cyclohexylammonium salt for 12 h at 37°C. Stained tissues were rinsed extensively in 70% (v v−1) ethanol to remove any residual chlorophyll.
Statistical analysis
A univariate analysis of variance (ANOVA) was performed using the Analysis ToolPak of Microsoft Excel 2000 (Microsoft Corporation).
Results and discussion
Efficient and reliable plant genetic transformation protocols depend on the performance of the underlying tissue culture and gene transfer systems. Plant regeneration, in particular, is of paramount importance in the development of an effective transformation protocol. Previously reported methods for the establishment of transgenic opium poppy plants have been based on direct shoot organogenesis (Park and Facchini 2000b) or somatic embryogenesis (Chitty et al. 2003) techniques. We used a systematic approach to facilitate A. tumefaciens-mediated gene transfer and plant regeneration using a commercial variety of opium poppy that was recalcitrant to published protocols. Somatic embryogenesis was selected over organogenesis due to the low efficiency of adventitious root formation on excised opium poppy shoots, but also to employ a tissue culture strategy that offers more potential to reduce the frequency of plants that escape selection (Park and Facchini 2000b). Chimeras are also eliminated due to the single-cell origin of somatic embryo. Proliferating callus was obtained from all parts of 7-day-old seedlings when cultured on a medium containing both auxin and cytokinin at a molar ratio between 1:1 and 1:2. The most prolific callus was obtained from cotyledons, followed by apical meristems and roots. However, callus produced by roots had the highest capacity for embryogenesis. Roots from 7-day-old seedlings were selected as the standard explant tissue for routine callus production. Once induced from pre-cultured explants the proliferating callus could be maintained indefinitely on a medium containing both auxin and cytokinin, or transferred to a medium containing a high level of auxin only for the efficient induction of embryogenic callus. Typically, 2–3 months were required to obtain vigorously proliferating, herbicide-resistant callus. After maintaining the callus for several months on a medium containing 10 mg mL−1 PPT approximately 75% of the total population of cultured cells were transgenic as estimated by histochemical staining for GUS activity (data not shown). The use of pre-cultured explants, rather than fresh-cut seedlings, substantially increased the frequency of herbicide-resistant embryogenic callus formation and, thus, substantially improved transformation efficiency. The embryogenic capacity of the original callus remained relatively stable over a 2-year period. The efficiency of embryogenic callus formation varied considerably among several cultivars tested (data not shown), suggesting that the establishment of callus primarily composed of white globular embryos is a major procedural component that requires empirical adjustment for different opium poppy cultivars. In the protocol of Chitty et al. (2003), transformation efficiency was variable, but transgenic plants were obtained for 14 of 15 tested genotypes, supporting the correlation between opium poppy genotype and susceptibility to A. tumefaciens infection and/or regeneration capacity.
Exposing the callus to a low density of A. tumefaciens for an extended period of time was also more effective than the more typical co-cultivation of explants with a higher density of bacteria. The possibility that Agrobacterium-mediated gene transfer is an ATP-dependent process (de la Riva et al. 1998) was taken into consideration by adding ATP and MgCl2 to the infection and co-cultivation media. Overall, the stable transformation frequency estimated by the histochemical staining of callus cultures for GUS activity was more reliable and efficient when all of these procedural refinements were combined. After the infection of pre-cultured explants with A. tumefaciens, the explants were returned to the callus induction medium supplemented with (1) the antibiotic timentin to prevent the re-growth of bacteria and (2) the herbicide PPT to suppress the proliferation of non-transformed tissues.
In opium poppy, somatic embryogenesis is a multi-step process. Embryogenic callus production was induced with the formation of white globular callus approximately 4 weeks after the transfer of homogenous proliferating callus to a medium containing 2,4-D. Various phytohormones and combinations thereof were initially tested with respect to the efficiency of embryogenic callus production (Fig. 1). High concentrations (2.5 and 5 μM) of 2,4-D produced the most consistent results, whereas the embryogenic response decreased with increasing concentrations of α-naphthaleneacetic acid (NAA). In contrast, cytokinins failed to cause a consistently positive effect. Subsequent to white globular callus formation, the induction of somatic embryogenesis required the transfer of embryogenic callus to a hormone-free medium to promote plantlet development (Fig. 2).

Effect of different auxins and cytokinins on the production of embryogenic callus from pre-cultured callus of opium poppy cultivar Louisiana. The medium consisted of B5 salts and vitamins, 1% (w v−1) glucose, 2% (w v−1) sucrose, 50 mM ATP, 50 mM MgCl2, 0.8% (w v−1) Difco noble agar, and the following phytohormones: 1–2.5 mM 2,4-D; 2–5 mM 2,4-D; 3–2.2 mM NAA; 4–4.4 mM NAA; 5–8.8 mM NAA; 6–4.4 mM NAA, 2.2 mM BA; 7–1 mM 2,4-D, 2.4 mM kinetin; 8–1 mM 2,4-D, 4.7 mM kinetin; 9–2.5 mM 2,4-D, 2.4 mM kinetin; 10–2.5 mM 2,4-D, 4.7 mM kinetin. Error bars represent the standard deviation of triplicate experiments. Bars with the same letter are not significantly different at P < 0.01 as determined by one-way ANOVA

Somatic embryogenesis from pre-cultured explants of opium poppy cultivar Louisiana and the conversion of somatic embryos into pre-rooted plantlets. a Formation of callus from pre-cultured root explants on medium containing 2.5 μM 2,4-D; b growth of white globular callus on medium containing 5 μM 2,4-D; c, d conversion of somatic embryos at the torpedo (c) and early cotyedonary (d) stages on hormone-free medium; e, f conversion of somatic embryos at the intermediate (e) and late (f) cotyledonary stages on hormone-free medium, g–i progressive stages in the development of opium poppy plantlets produced from somatic embryos on hormone-free medium. Plantlets were transferred to soil after the induction of roots
Since PPT substantially decreased the conversion rate of globular callus and caused abnormal embryo development in the opium poppy cultivar Louisiana, the selection agent was removed from the hormone-free somatic embryo induction medium. An advantage of somatic embryogenesis compared with organogenesis as the regeneration system for opium poppy is the opportunity to aggressively apply the selection agent at the callus induction and callus proliferation stages, and remove the herbicide during somatic embryogenesis. This uncoupling of selection and regeneration resulted in the achievement of a reasonable transformation frequency without a substantial increase in the number of plants that escaped selection. The proportion of escapes averaged approximately 35%.
Phosphinothricin inhibits glutamine synthetase activity, which restricts the availability of L-glutamine at the somatic embryo induction stage. Addition of L-glutamine to the culture medium was shown to improve the embryogenic regeneration efficiency of certain plant species (Sobha et al. 2003; Jin et al. 2005), suggesting a role for L-glutamine during somatic embryogenesis. Generally, the regeneration frequency of opium poppy embryogenic cultures was considerably reduced when PPT was added to the medium. The regeneration efficiency of PPT-resistant embryogenic callus was similar to that of wild type embryogenic callus cultured when L-glutamine was added to the medium.
Developing embryos appeared after 4–6 weeks on hormone-free medium. Somatic embryos typically grew in clusters joined at their base, and lacked roots. To induce root formation, embryos at the cotyledonary stage were transferred to a hormone-free medium lacking organic supplements. Rooting has been the major limiting factor affecting the efficiency of all reported opium poppy transformation and regeneration systems (Nessler 1982; Park and Facchini 2000b; Chitty et al. 2003). However, certain conditions and treatments were found to substantially improve rooting efficiency including: (1) separating embryo clusters into individual embryos, (2) preventing direct contact between embryos and the agar medium by placing sterilized filter paper placed on the agar surface, (3) briefly (i.e. 2 to 5 h) exposing the embryos to a high concentration (i.e. 500 μM) of IBA, and (4) using relatively cool (i.e. 20°C day and 15°C night) cultivation temperatures. Plantlets with roots were transferred to soil and grown under clear plastic covers to maintain relatively high humidity for 7 days. Subsequently, plantlets were cultivated under greenhouse conditions. A flowchart summary of the complete transformation protocol is shown in Fig. 3.

Flow chart summary of the Agrobacterium-mediated genetic transformation and regeneration of opium poppy plants
The expression of pat and gus transgenes was determined in T1 progeny from seven different regenerated opium poppy lines by measuring the relative abundance of pat and gus gene transcripts and corresponding enzyme activities (Fig. 4). Real-time PCR showed the accumulation of pat and gus mRNAs in the leaves of all regenerated lines demonstrating the selection efficiency of the reported transformation protocol. In contrast, pat and gus transcripts were not detected in wild type opium poppy leaves. Similarly, PAT and GUS enzyme activities were also detected in the leaves of all regenerated lines, but not in wild type plants. In general, direct correlations between mRNA and enzyme activity levels were inconsistent for both transgenes possibly due to the effects of transgene copy number, the location of chromosomal insertion, or post-transcriptional gene-silencing (Baulcombe 1996). Cytohistochemical localization of GUS activity showed that the transformation protocol resulted in fully transgenic T1 plants (Fig. 5). The CaMV 35S promoter-GUS fusion contained in the pCAMBIA3301 binary vector was expected to result in constitutive GUS activity in all cell types. GUS activity was observed in roots and all shoot organs, but was substantially higher in the outer stem cortex compared with the pith (Fig. 5). In contrast, GUS activity was not detected in any wild type opium poppy tissues (data not shown). The detection of transgene mRNAs and corresponding enzyme activities in all organs of T1 progeny shows the genomic integration and inheritance of the T-DNA from pCAMBIA 3301.

Relative phosphinothricin acetyltransferase (PAT) (a, b) and β-glucuronidase (GUS) (c, d) gene transcript (a, c) and enzyme activity (b, d) levels in the leaves of wild type and seven transgenic lines of opium poppy. Total RNA for real-time PCR was isolated from the leaves of T1 plants. Real-time PCR was performed using primers specific to either the PAT or GUS coding regions. Expression levels were normalized against transcripts from a constitutively expressed opium poppy elongation factor 1a gene. PAT activity was determined by measuring the transfer of the radioactive label from [acetyl-1-14C]-acetyl coenzyme A to phosphinothricin. GUS activity was assayed by measuring the formation of 4-methylumbelliferone from 4-methylumbelliferyl-β-d-glucuronide using fluorescence spectroscopy. Error bars represent the standard deviation of triplicate experiments. Bars with the same letter are not significantly different at P < 0.01 as determined by one-way ANOVA

Histochemical localization of β-glucuronidase (GUS) activity in T1 opium poppy plants. The T1 seeds were obtained from plants regenerated from phosphinothricin-resistant embryogenic callus transformed with cauliflower mosaic virus (CaMV) 35S promoter-phosphinothricin acetyltransferase (pat) selectable marker gene and 35S promoter-gus reporter gene fusions. a Stem and leaves, b cross-section of a mature stem, c roots, d cross section of a mature root
We report several innovations in the development of effective genetic transformation and regeneration protocols for the establishment of herbicide-resistant opium poppy plants. The commercial variety used was recalcitrant to other published protocols (Park and Facchini 2000b; Chitty et al. 2003). The availability of the reliable transformation protocol described in this study will facilitate the alteration of the agronomic characteristics and alkaloid content of opium poppy crops (Allen et al. 2004; Frick et al. 2004; Larkin et al. 2007).
Abbreviations
- BA:
-
6-Benzyladenine
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- CaMV:
-
Cauliflower mosaic virus
- IBA:
-
Indolebutyric acid
- GUS:
-
β-Glucuronidase
- MUG:
-
4-Methylumbelliferyl-β-d-glucuronide
- NAA:
-
α-Naphthaleneacetic acid
- PAT:
-
Phosphinothricin acetyltransferase
- PPT:
-
Phosphinothricin, also known as glufosinate ammonium
- TLC:
-
Thin-layer chromatography
References
Allen RS, Millgate AG, Chitty JA, Thisleton J, Miller JAC, Fist AG, Gerlach WA, Larkin PJ (2004) RNAi-mediated replacement of morphine with the non-narcotic alkaloid reticuline in opium poppy. Nat Biotechnol 22:1559–1566
Auriac MC, Timmers AC (2007) Nodulation studies in the model legume Medicago truncatula: advantages of using the constitutive EF1alpha promoter and limitations in detecting fluorescent reporter proteins in nodule tissues. Mol Plant Microbe Interact 20:1040–1047
Baulcombe DC (1996) RNA as a target and initiator of post-transcriptional gene silencing in transgenic plants. Plant Mol Biol 32:79–88
Bradford MM (1976) A rapid and sensitive method for quantification of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem 72:512–516
Chitty JA, Allen RS, Fist AJ, Larkin PJ (2003) Genetic transformation in commercial Tasmanian cultivars of opium poppy, Papaver somniferum, and movement of transgenic pollen in the field. Funct Plant Biol 30:1045–1058
de la Riva GA, González-Cabrera J, Vázquez-Pardón R, Ayra-Pardo C (1998) Agrobacterium tumefaciens: a natural tool for plant transformation. Electron J Biotechnol 1:119–133
Dieu P, Dunwell JM (1988) Anther culture with different genotypes of opium poppy (Papaver somniferum): effect of cold treatment. Plant Cell Tissue Organ Cult 12:263–271
Dubedout M (1993) Analysis of progenies from a circular plan of crosses in poppy (P. somniferum L.), PhD Thesis, University of Paris, Orsay, France
Frick S, Chitty JA, Kramell R, Schmidt J, Allen RS, Larkin PJ, Kutchan TM (2004) Transformation of opium poppy (Papaver somniferum L.) with antisense berberine bridge enzyme gene (anti-bbe) via somatic embryogenesis results in an altered ratio of alkaloids in latex but not in roots. Transgenic Res 13:607–613
Gamborg OL, Miller RO, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Hagel J, MacLeod BP, Facchini PJ (2007) Opium poppy. In: Pua EC, Davey MR (eds) Biotechnology in agriculture and forestry: tropical crops II. Springer, Heidelberg, pp 169–187
Ilahi I, Ghauri EG (1994) Regeneration in cultures of Papaver bracteatum as influenced by growth hormones and temperature. Plant Cell Tissue Organ Cult 38:81–83
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Jin S, Zhang X, Liang S, Nie Y, Guo X, Huang C (2005) Factors affecting transformation efficiency of embryogenic callus of Upland cotton (Gossypium hirsutum) with Agrobacterium tumefaciens. Plant Cell Tissue Org Cult 81:229–237
Kassemi MA, Jacquin A (2001) Somatic embryogenesis, rhizogenesis, and morphinan alkaloids production in two species of opium poppy. J Biomed Biotechnol 1/2:70–78
Kosugi S, Ohashi Y, Nakajima K, Arai Y (1990) An improved assay for β-glucuronidase in transformed cells: methanol almost completely suppresses a putative endogenous β-glucuronidase activity. Plant Sci 70:133–140
Larkin PJ, Miller JAC, Allen RS, Chitty JA, Gerlach WL, Frick S, Kutchan TM, Fist AJ (2007) Increasing morphinan alkaloid production by over-expressing codeinone reductase in transgenic Papaver somniferum. Plant Biotechnol J 5:26–37
Le Flem-Bonhomme V, Laurain-Mattar D, Fliniaux MA (2004) Hairy root induction of Papaver somniferum var. album, a difficult-to-transform plant, by A. rhizogenes LBA 9402. Planta 218:890–893
Levy A, Milo J (1997) Genetics and breeding of Papaver somniferum. In: Bernáth J (ed) Poppy: the genus Papaver. Harwood Academic, Amsterdam, pp 93–103
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Nessler CL (1982) Somatic embryogenesis in opium poppy, Papaver somniferum. Physiol Plant 55:453–458
Nessler CL (1998) In vitro culture technologies. In: Bernath J (ed) Poppy: the genus Papaver. Harwood Academic, Amsterdam, pp 209–218
Ovecka M, Bobak M, Blehova A, Kristin J (1997) Papaver somniferum regeneration by somatic embryogenesis and shoot organogenesis. Biol Plant 40:321–328
Ovecka M, Bobak M, Samaj J (2000) A comparative structural analysis of direct and indirect shoot regeneration of Papaver somniferum L. in vitro. J Plant Physiol 157:281–289
Park SU, Facchini PJ (2000a) Agrobacterium rhizogenes-mediated transformation of opium poppy, Papaver somniferum L., and California poppy, Eschscholzia californica Cham., root cultures. J Exp Bot 51:1005–1016
Park SU, Facchini PJ (2000b) Agrobacterium-mediated transformation of opium poppy, Papaver somniferum L., via shoot organogenesis. J Plant Physiol 157:207–214
Park S-U, Facchini PJ (2000c) High efficiency somatic embryogenesis and plant regeneration in California poppy, Eschscholzia californica Cham. Plant Cell Rep 19:421–426
Park S-U, Facchini PJ (2001) Somatic embryogenesis from embryogenic cell suspension cultures of California poppy, Eschscholzia californica Cham. In Vitro Plant Cell Dev Biol 37:35–39
Sagare AP, Lee YL, Lin TC, Chen CC, Tsay HS (2000) Cytokinin-induced somatic embryogenesis and plant regeneration in Corydalis yanhusuo (Fumariaceae)—a medicinal plant. Plant Sci 160:139–147
Schuchmann R, Wellmann E (1983) Somatic embryogenesis of tissue cultures of Papaver somniferum and Papaver orientale and its relationship to alkaloid and lipid metabolism. Plant Cell Rep 2:88–91
Singh SP, Khanna KR (1991) Genetic variability for some traits in opium poppy (Papaver somniverum L.). Narendra Deva J Agric Res 6:88–92
Sobha S, Sushamakumari S, Thanseem I, Kumari Jayasree P, Rekha K, Jayashree R, Kala RG, Asokan MP, Sethuraj MR, Dandekar AM, Thulaseedharan A (2003) Genetic transformation of Hevea brasiliensis with the gene coding for superoxide dismutase with FMV 34S promoter. Curr Sci 85:1767–1773
Trieu AT, Harrison MJ (1996) Rapid transformation of Medicago truncatula: regeneration via shoot organogenesis. Plant Cell Rep 16:6–11
Wakhlu AK, Bajwa PS (1986) Regeneration of uniform plants from somatic embryos of Papaver somniferum (opium poppy). Phytomorphology 36:101–105
White PR (1967) Plant cell and tissue culture. In: Wilt FH, Wessels NK (eds) Methods in developmental biology. Thomas Y. Crowell, New York, pp 555–564
Acknowledgments
We thank Katy Nour and David Bird for technical assistance. Seeds of the Louisiana opium poppy cultivar were a kind gift from Sanofi-Aventis (France). This work was financed through an industrial research contract with Sanofi-Aventis. P.J.F. holds the Canada Research Chair in Plant Metabolic Processes Biotechnology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Debergh.
Rights and permissions
About this article
Cite this article
Facchini, P.J., Loukanina, N. & Blanche, V. Genetic transformation via somatic embryogenesis to establish herbicide-resistant opium poppy. Plant Cell Rep 27, 719–727 (2008). https://doi.org/10.1007/s00299-007-0483-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-007-0483-8