
Abstract
In order to visualize the specific state of tobacco pollen undergoing dedifferentiation from immature pollen to embryogenic cells, we established tobacco marker lines transgenic for a vital reporter gene regulated under the transcriptional control of an 840 bp fragment, named A22pro. This fragment was obtained from the 5′-flanking region of a gene corresponding to a cDNA named A22 that was previously isolated through differential screening from a cDNA library prepared from tobacco pollen undergoing dedifferentiation. The reporter gene, named H3sGFP, consisting of synthetic green fluorescent protein gene (sGFP) and tobacco H3 histone gene for nuclear localization, was designed to distinguish the gene expression in the generative cell from that in the vegetative cell in a pollen grain. The marker line produced pollen showing a green fluorescent signal in the generative nuclei (GN) but the expression level of the transgene was low. Pollen after culture for dedifferentiation showed an intense signal transiently in the vegetative nuclei (VN), at a specific developmental stage of pollen, with a rapid increase of expression level of the transgene. Serial observations revealed that all androgenic embryos originated from the pollen grains that had shown the signal in their VN. Thus, A22pro is originally functional in gametogenesis but is activated in VN of pollen undergoing embryogenic dedifferentiation. Additionally, we observed a gene expression pattern identical to that described above, using another 5′-flanking region of a gene for a cDNA, named B27pro, homologous to A22 as a promoter of the reporter gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the first observation by Guha and Maheshwari (1964, 1966), embryogenesis from microspore/pollen through anther culture has been reported in many plant species. The resulting androgenic plants are haploids in principle and can be fertile diploids through spontaneous or artificial duplication of chromosomes. Such doubled haploids seem to be identical to homozygous plants. Therefore, embryogenesis from microspore/pollen can be used to shorten the years and to decrease the labor for producing pure lines in breeding work. In fact, many successful examples have been reported in tobacco, rice, wheat, and other crops (see Jain et al. 1996). However, popularization of the breeding method using androgenic plants is not easy because the frequency of androgenesis is too low for actual work. A molecular biological approach may help to raise the frequency of androgenesis in future but the induction mechanism of the phenomenon is entirely unknown (see Reynolds 1997; Pechan and Smykal 2001).
Tobacco is a model plant in which the androgenic response can be induced at a high frequency by anther and pollen culture methods. Early histological observations of anthers in culture suggested that tobacco pollen embryos originated from vegetative cells of bicellular pollen grains (Dunwell and Sunderland 1974). Kyo and Harada (1986) reported that immature tobacco pollen at the mid-bicellular stage (Stage III) could be induced to undergo the conversion process in vitro, so-called embryogenic dedifferentiation, from immature pollen to embryogenic cells. When Stage III pollen was cultured under a condition lacking a suitable nitrogen source necessary for maturation the dedifferentiation was induced at a high frequency. The immature pollen younger or older than Stage III, i.e., early or late bicellular stage (Stage II or IV, respectively) could not be dedifferentiated by the basic culture but we could induce dedifferentiation of Stage IV pollen using an acidified medium (Kyo 1990). Touraev et al. (1996) reported another unique culture method for inducing embryos from tobacco microspores by combining a starvation treatment and a high temperature treatment.

Tobacco BY-2 cells transgenic for 35S/H3sGFP in the stationary phase a, e, in the mitotic phase b, f and soon after cell division c, d, g, h. a–d are fluorescent images and e–h are their phase-contrast images, respectively. c and d show the same area in different foci. Bar = 5 μm
Sixteen species of cDNAs have been cloned as candidates for the marker of pollen dedifferentiation through differential screening and gene expression analyses (Kyo et al. 2003). However, it was not clear whether the resulting androgenic embryos originated from the cells that expressed the marker genes. We intended to isolate the promoters of genes for the marker cDNAs and to visualize their transcriptional activity in vegetative nuclei (VN) and generative nuclei (GN) by using a vital reporter gene. The 5′-upstream flanking regions of genes for the marker cDNAs were isolated by inverse PCR, linked to the 5′-end of a green fluorescent protein gene and integrated into a binary vector for Agrobacterium-mediated gene transfer. Through serial observation of cultured pollen isolated from the plants transgenic for the reporter gene under the transcriptional control of a promoter, it seems possible to estimate whether the promoter is activated in the pollen grains undergoing the dedifferentiation. As a reporter gene, we designed a fusion gene, named H3sGFP, consisting of synthetic green fluorescent protein (sGFP) gene (Chiu et al. 1996) and tobacco H3 histone gene (accession number AB032544) isolated from a cDNA library previously established (Kyo et al. 2000) to locate the fusion gene products in nuclei. The design for nuclear localization allowed us to detect the fluorescent signal in a generative cell without being effaced by the signal in the cytoplasm of the vegetative cell. Additionally, the concentration of the fluorescent protein on the nuclei should increase the intensity of the fluorescent signal.
Two cDNA clones, named B27 (Kyo et al. 2003) and A22, were previously isolated as marker genes for the pollen dedifferentiation. Their nucleotide sequences showed 92% identity with each other in whole sequences. The expression of the gene for B27 was highly associated with the pollen dedifferentiation and was rapidly increased to the maximum level within a day after the start of the pollen culture (Kyo et al. 2003). In a BLAST search (Altschul et al. 1997) their putative translational products showed similarity (33% in 250 amino acids) in part with a putative protein named potato late blight resistance protein-like (accession no. AAU89785, Ballvora et al. unpublished) but their functions remain unknown. In this report, we describe the feature of two promoters for A22 and B27 and a gene expression pattern conferred by the promoters in the pollen undergoing pollen maturation and dedifferentiation.
Materials and methods
Construction of pBI-35S/H3sGFP
cDNA for tobacco H3 histone was isolated from a cDNA library previously established (Kyo et al. 2000) and subcloned into pBluescript SK(−) through an in vivo excision system (Stratagene). The coding region was amplified by PCR using a pair of primers, 5′-gggtcgaccATGAGCACTGTCCAGAACACACC and 5′-gggcaccatggAGACTCTTCTTTTCCCTTCGGCTTC (capitals show H3 histone-coding region and small letters show the sequence giving SalI or NotI site). The resulting fragment was treated with SalI and NotI and then in-frame inserted into the SalI-NotI site in a plasmid, pTH-2 (Chiu et al. 1996) harboring the engineered and synthesized GFP gene, named sGFP (S65T). The resulting plasmid was digested with HindIII and EcoRI to generate a fragment consisting of the three regions, CaMV35S promoter (approximately 400 bp), a coding region for a fusion protein referred to as H3sGFP, and the Nos terminator. This fragment was inserted into the HindIII-EcoRI site of the binary vector, pBI121 (Clontech, Palo Alto, CA, USA). The resulting plasmid was referred to as pBI-35S/H3sGFP and used for transformation of the tobacco BY-2 cell line to observe the intracellular localization of H3sGFP products (Fig. 1).

Alignments of the nucleotide sequences of 5′-upstream regions (A22pro and B27pro) of the genes for A22 and B27 clones and a MAR, pS206-1 a and the construct of transgene in the binary vector used for generating plants transgenic for A22pro/H3sGFP b. Capitals: 128 bp of the 5′-end of cDNA sequence for A22. Asterisks: consistent sites among A22pro, B27pro, and pS206-1. Double underline: putative TATA box. Open box: a complementary sequence of GTGA motif. Single underline:ARR1 binding motif (NGATT) or its complementary sequence (AATCN)
Isolation of promoters
Nicotiana tabacum cv. Samsun genomic DNA was prepared from seedlings by the method described by Liu et al. (1995), digested with PstI, treated with T4 DNA ligase (New England Biolabs, Beverly, MA, USA) to generate circular DNAs and then used as a template in inverse PCR (Triglia et al. 1988). Two pairs of primers, 5′-GAACTCTGACAACCTCGAACTC and 5′-TTGGAATGGTTGTCGCTACTTGA for the 1st PCR, 5′-ACGAAATCCCGAACTCCGTCTT (Primer 2 in Fig. 2a) and 5′-TGAGGTAGATCCGGGTTATGCT for the 2nd PCR, were designed from the A22 cDNA sequence. After the 1st PCR, an aliquot of the reaction mixture was diluted with 500-fold volume of distilled water and 1 μl of the dilution was added to 20 μl of the 2nd PCR mixture. The amplified fragment (approximately 1.8 kbp) was cloned directly into pPCR-Script Amp SK(+) (Stratagene, La Jolla, CA, USA) at the EcoRV site modified with a 5′-T overhang and was sequenced as described previously (Kyo et al. 2000). The authentic DNA sequence corresponding to the amplified fragment was presumed by comparing the sequences of several clones amplified independently and a clone with the identical sequence was selected. The resulting plasmid was cut with SpeI, blunted with T4 DNA polymerase, and then cut again with SalI to generate a 840 bp fragment, referred to as A22pro (Fig. 2a) containing 128 bp of the noncoding region of A22 cDNA and its 5′-upstream. This fragment replaced the CaMV35S promoter region in pBI-35S/H3sGFP to generate a chimeric gene referred to as A22pro/H3sGFP. pBI-35S/H3sGFP treated with HindIII were blunted with Klenow enzyme and then cut again with SalI. The linearized plasmid with SalI site and blunt end was ligated with A22pro (840 bp). Figure 2b shows the T-DNA region in the resulting plasmid referred to as pBI-A22pro/H3sGFP.
Based on the B27 cDNA sequence two pairs of primers, 5′-CAAGAAAACGAACGAAGACAGCA and 5′-ATCTCCGACATCACCCCAAGTT for the 1st PCR, 5′-CCGGATCCGAAACTCGAAATCG and 5′-TTGGAATGGTTGTCGCTACTTGA for the 2nd PCR, were designed for inverse PCR using tobacco genomic DNA digested with SphI and treated with T4 DNA ligase. The amplified fragment (approximately 3 kbp) was cloned into pPCR-Script Amp SK(+) and the authentic sequence of the 5′-upstream region of B27 gene was confirmed as described above. A pair of primers, 5′-aagctTGTGAACAACTAATTTTTTACCACA and 5′-tctcgAGTTCTCTGTGGAATATAATCAATG (small letters show the sequence giving HindIII or XhoI site) were designed to amplify B27pro sequence (Fig. 2a). The amplified fragments were ligated to pBI-35S/H3sGFP at HindIII-SalI site after being treated by HindIII and XhoI. A clone with no nucleotide difference from the authentic sequence was selected and used for transformation of tobacco.

Expression analysis of the gene for the A22 clone in microspore/pollen and somatic tissues. Seedlings 1 and 2 were harvested just after germination and just after cotyledons expanded, respectively. Root, stems, and leaves were obtained from 1-month-old plants. The stems contain some nodes and petioles. Anthers were obtained from flower buds with the indicated corolla length, respectively. Pollen was harvested after the indicated period (h) from the start of culture. Clone B38 (accession number: AB032545) was used an internal standard as described previously (Kyo et al. 2003)
Plant transformation
Agrobacterium tumefaciens LBA4404 transformed with pBI-A22pro/H3sGFP or pBI-B27pro/H3sGFP was used for transformation of tobacco leaf disks by a method reported previously (Horsch et al. 1985). Transformants were screened on MS medium (Murashige and Skoog 1962) supplemented with 0.8% agar, kanamycin (200 mg l−1), carbenicilin (200 mg l−1), 5 μM benzyladenine, and 1 μM NAA. Shoots generated from leaf disks on the medium were cut and replaced on hormone-free MS agar medium with kanamycin (200 mg l−1) and carbenicilin (200 mg l−1). Regenerants with roots were replanted in soil in a pot and grown in a closed phytotron regulated at 20–25°C under a natural light condition. The transgene in the genome of regenerants was detected by PCR using the pair of primers noted below.
Cell culture and observation
Tobacco transformants possessing A22pro/H3sGFP homozygously were selected from offspring (T2) of a regenerant (T1) and one of them was used as a pollen donor. The pollen donor plants were cultivated in a closed phytotron. Preparation of pollen population and basic culture method were as reported previously (Kyo and Harada 1986) except that the second culture step was omitted and the basal medium was modified to medium F (200 mM mannitol, 100 mM maltose, 2 mM KCl, 1 mM K2SO4, 0.5 mM KH2PO4, 0.5 mM K2HPO4, pH 6.8). Pollen prepared from flower buds with a corolla length of 15–17, 20–22, and 26–28 mm are referred to as pollen of Stage II, III, and IV, respectively. For serial observation, pollen was embedded in droplets of medium F with 0.5% (w/v) of low gelling temperature agarose (SeaPlaque, FMC, Rockland, ME, USA) on chambered cover glass (Lab-Tek chambered cover glass, No.136439, Nunc, Denmark) and observed under an inverted fluorescent microscope (IMT2, Olympus, Tokyo, Japan). The time for observing under a fluorescent microscope was kept as short as possible to avoid fatal damage to pollen by excitation light. Pollen viability was lowered by the application of laser to pollen for the observation by confocal laser scanning microscopy. Therefore, we used a fluorescent microscope for the observation of pollen.
BY-2 cells were cultured as described previously (Kyo et al. 2000) and observed by confocal laser scanning microscopy (Leica Microsystems, Wetzlar, Germany).
Expression analysis
We examined the level of specific mRNA by RT-PCR as reported previously (Kyo et al. 2000). First stranded cDNA population was prepared from the total RNA isolated from each cell population and was added to a PCR mixture with a pair of primers specific to the target sequence. The sequence was amplified to obtain a detectable amount of the target sequence, but the number of amplification cycles was limited to approximately 20. The amplified DNA in each sample was separated by electrophoresis, transferred to a membrane, and visualized with a DIG- labeled specific probe and detection system. The sequences of specific primer sets for the coding region of A22, sGFP, and B38 were TTGGAATGGTTGTCGCTACTTGA and GGCCTTCTCATTTAGCTGTACG, ACGGCAAGCTGACCCTGAAGT and GACCATGTGATCGCGCTTCTC, and GAGAAGGTTGTGCAGGAGAAGA and GGCCTTTGGTACATTGGCATTC, respectively.
Results and discussion
Expression analysis of A22 clone
Figure 3 shows the expression level of the gene for A22 in somatic tissues and microspores/pollen. The expression pattern was similar to that for B27 shown previously by Kyo et al. (2003). Seedlings, root, stems with some nodes, and leaves contained undetectable levels of the transcripts. Fresh anthers including microspores/pollen at various developmental stages contained a low level of the transcripts. Pollen at the early, mid-, and late bicellular stages, Stages II, III, and IV, respectively, was cultured for 48 h in medium F. Embryogenic cell division was observed only in Stage III pollen after 3 weeks of culture. The level of the transcript in Stage III pollen increased markedly 15 h after the start of culture and the level was kept high for 48 h. A much lower level was observed in the younger or older pollen (pollen at Stage II and IV). In addition, ovules and styles obtained from flower buds of various lengths did not have a detectable level of the transcripts (data not shown).
Cloning of 5′-upstream regions of the genes for A22 and B27
In order to obtain new insight into the molecular mechanism of the embryogenic dedifferentiation of tobacco immature pollen, we isolated an approximately 1600 bp 5′-upstream region of the genes corresponding to A22 by inverse PCR. The 1600 bp and 840 bp (SpeI-SalI fragment, Fig. 2a) fragments were linked to the 5′-end of H3sGFP gene in a vector (Fig. 2b) to generate tobacco transformants as described later. Using the transgenic pollen generated by the transformants possessing the 1600 or 840 bp fragment as the promoter of the transgene, we obtained identical results in the expression analysis of the reporter gene and in the fluorescent microscopic observation (data not shown). Therefore, we focused on the shorter fragment of 840 bp, named A22pro, in this report.
Interestingly, A22pro (Fig. 2a) possesses an approximately 300 bp sequence in the region from −366 to −641 (counted from the position corresponding to 5′-end of the cDNA of A22) that showed 74% identity in 306 bp with a tobacco genomic sequence, named pS206-1 (Michalowski et al. 1999). pS206-1 is a tobacco genomic sequence co-purified with nuclear matrixes prepared by a method involving lithium diiodosalicylate, and seems to be one of tobacco nuclear matrix attachment regions (MARs). MARs are DNA fragments that bind to the nuclear matrix and form the anchor points of loop domains in chromatin. The AT-rich DNA fragments are supposed to be concerned with DNA replication and transcriptional regulation of gene including cooperative effects with enhancer or silencer and stabilization of transgene expression by shielding from position effect (see Bode et al. 1995, 1996).
We isolated the 5′-upstream region of the gene corresponding to B27, named B27pro. As shown in Fig. 2a, B27pro also possesses an MAR-like sequence in the region from −650 to −350 (counted from the 5′-end of the B27 cDNA sequence). In this region, the similarity between A22pro and B27pro is very high (74% identity in 350 bp). However, in the downstream region, the similarity between the two was low (54% identity in 250 bp). Thus, the MAR-like sequence was commonly found in A22pro and B27pro as a conservative region, suggesting that the sequence plays an important role in their transcriptional regulation specific to the dedifferentiation of pollen. Using heterozygous plants transgenic for B27pro/H3sGFP we confirmed that B27pro also conferred a gene expression pattern with resulting appearance of the reporter protein in a manner identical to that of A22pro.
Using the PLACE database program (Higo et al. 1999), we searched for regulatory elements common in A22pro and B27pro. TATA boxes, TATAAA and TATATATA, were found in the region from −60 to −65 (counted from the position corresponding to 5′-end of cDNA) in A22pro and in the region from −67 to −76 in B27pro, respectively. An Arabidopsis response regulator homolog 1 (ARR1) binding motif, NGATT, or its complementary sequence were found in six and five sites in A22pro and B27pro, respectively (Fig. 2a). ARR1 seems to be a transcription factor regulated by a cytokinin signal transduction system (Sakai et al. 2000). Four and six repeats of TCAC were found in A22pro and B27pro, respectively (Fig. 2a). The complementary sequence of TCAC, i.e., GTGA is a cis-regulatory element found in the promoter of a tobacco gene named g10 whose expression is specific to pollen maturation (Rogers et al. 2001) though the function of g10 products is unknown.
Intracellular localization of H3sGFP in BY-2 cells
Nuclear localization of fusion gene product consisting of a fluorescent protein and a histone H2A was previously used to visualize intact nuclei also in plant cells (e.g., Boisnard-Lorig et al. 2001). However, it was not clear whether the tobacco cDNA coding for a histone H3 homolog is available for nuclear localization of the vital reporter protein. To examine the accurate localization of the fusion gene product (H3sGFP) in tobacco cells, we transformed BY-2 cells with a binary vector pBI-35S/H3sGFP. The resulting transgenic BY-2 cells showed green fluorescence in their nuclei or mitotic chromosomes (Fig. 1a and b), indicating that H3sGFP is located like the natural H3 histone.
In Fig. 1a, c, and d, nucleoli did not show any fluorescent signals but were visualized clearly. The difference in the number and size of nucleoli was clear between the actively proliferating phase (Fig. 1c and d) and the stationary phase (Fig. 1a). The fusion reporter gene, H3sGFP, should be a clue to monitor the change of intranuclear structure associated with the cell cycle and physiological state.
Tobacco plants transgenic for A22pro/H3sGFP
Using a binary vector, pBI-A22pro/H3sGFP, we transferred the chimeric reporter gene to tobacco genome. We obtained two transformants (T1) generating mature pollen with green fluorescence in their generative cells at the frequency of approximately 50%, indicating that these pollen donors possess a single functional transgene in their each genome. A homozygous line was obtained from the T2 generation of one of the two transformants and used for the pollen donor to examine the expression manner of A22pro/H3sGFP. We observed no fluorescent signal in the nuclei of somatic cells in shoots, stems, leaves, and roots of those transformants, and no evident abnormality in fertility and morphology in the life cycle.

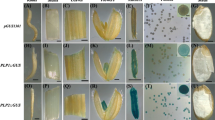
Tetrad microspores and pollen grains isolation from anthers of homozygous plant transgenic for A22pro/H3sGFP. Tetrad microspores a, f, Stage II pollen b, g, Stage III pollen c, h, Stage V pollen d, i, and mature pollen e, j were isolated from flower buds with a corolla length of 6, 15, 20, 35, and 52 mm, respectively. f–j are fluorescent microscopic images of a–e, respectively. k A pollen tube growing in medium C (Kyo and Harada 1986): an overlay image by fluorescence (green) and transmitted light (gray-scale) using confocal microscopy. Bar = 50 μm

Aspects of pollen grains cultured for 30 h. a, b, and d originated from pollen at Stages II, III, and IV cultured in medium F, respectively. c is from Stage III pollen cultured in medium F supplemented with 3 mM glutamine. e–h are the fluorescent microscopic images of a–d, respectively. i is a magnified fluorescent image from Stage III pollen cultured in medium F. Note the difference in diameter between VN and GN. Bar = 50 μm
Appearance of H3sGFP in pollen maturation
Figure 4 shows the aspects of pollen at various developmental stages freshly isolated from the homozygous line transgenic for A22pro/H3sGFP. The cell wall (exine) of microspore/pollen older than tetrad (Fig. 4a and f) showed yellow-green autofluorescence under a fluorescent microscope, but the fluorescent signal originating from H3sGFP was greener than the autofluorescence from exine. The nuclei with the fluorescent signal shown in Fig. 4h, i and j were GN judging from the size and the peripheral location in the pollen. As shown in Fig. 5i, the diameter of GN and VN were approximately 6 and 12 μm, respectively. The signal was observed in GN from just after Stage II to the mature stage but not in VN (Fig. 4h–j). In mature pollen, GN showed a spindle shape (Fig. 4j). The fluorescence in GN was maintained when GN was passing through the growing pollen tube (Fig. 4k), but no sperm nuclei could be found. Whether or not the products of A22 gene accumulate in GN during the pollen developmental process remains to be confirmed.
Recently a promoter of a generative cell-specific gene (LGC1) was identified by the transient assay using GFP gene and by cytochemical assay using a β-glucuronidase gene in a stable transformant (Singh et al. 2003). However, neither morphological change nor behavior of intact GN in living pollen was observed in their report. Therefore, the present result is the first successful example to visualize intact GN at a high frequency by stable expression of a fluorescent protein gene. The chimeric gene (A22pro/H3sGFP) seemed to be useful for observing the nuclear morphology in the gametogenesis.
Appearance of H3sGFP in pollen dedifferentiation
We cultured the pollen in a medium for inducing dedifferentiation. An intensive green fluorescent signal appeared in the VN and the frequency of such VN clearly differed with the developmental stage of pollen (Fig. 5). The Stage III pollen had a much higher percentage of VN with the signal (approximately 95%, Fig. 5f) than the Stage II (Fig. 5e) or Stage IV pollen (Fig. 5h). The Stage II pollen showed a weak signal only in GN but not in VN (Fig. 5e). In the Stage IV pollen population (Fig. 5h), a few pollen grains showed fluorescence in their VN, but they seemed to have originated from relatively younger pollen (Stage III) mingled into this population judging from the morphological feature of their cytoplasm (Fig. 5d) where VN appears surrounded by a circle. This feature is a morphological marker of the dedifferentiation as previously described by Kyo and Harada (1986). Stage III pollen cultured in medium F supplemented with 3 mM glutamine, matured and the intensity of fluorescence in VN was much lower than that cultured in the absence of glutamine (compare Fig. 5g with f). The highest frequency of pollen with fluorescent VN was observed in the Stage III pollen cultured in the absence of glutamine. This condition is consistent with that for inducing the embryogenic dedifferentiation of pollen. The fluorescent signal in GN was maintained in a pollen undergoing dedifferentiation (Fig. 5i).

Expression analysis of the transgene (A22/H3sGFP) and the gene for A22. Total RNA was prepared from pollen at Stages II, III, and IV cultured in medium F with or without 3 mM glutamine for indicated period (h)

Serial observation of pollen grains transgenic for A22/H3sGFP during culture. All aspects show the same area 2 h a, e, 30 h b, f, 75 h c, g, and 21 days d, h after the start of culture. e–h are the fluorescent images for a–d, respectively. Red arrows indicate an example of pollen that showed green fluorescence transiently in its VN and then developed to EM. Blue arrows indicate an example of pollen that showed no fluorescence in VN and then died without cell division. Bar = 50 μm
Expression level of A22pro/H3sGFP gene and A22 gene
Is the appearance of the fluorescent signal from H3sGFP coincident with the expression of the transgene, A22pro/H3sGFP or the expression of the endogenous gene for A22? The levels of the transcripts for H3sGFP and A22 gene during the early period of culture were monitored by RT-PCR (Fig. 6). The transgene and A22 gene were expressed in an almost identical pattern in Stage III and Stage IV pollen but not in Stage II pollen. This suggests that A22pro in the transgene and the endogenous promoter of A22 gene were under the same transcriptional control at least in both Stage III and Stage IV pollen even when cultured in the presence of glutamine. The level of the transcripts increased with culture time until 49 h in the culture of Stage III pollen, and for only 17 h followed by a decrease to a trace level after 49 h in the culture of Stage IV pollen. This difference in the expression pattern is consistent with the difference in the frequency of fluorescent VN between the Stage III and Stage IV pollen populations (Fig. 5f and h).
In the cultured Stage II pollen, the transcription level of the transgene was higher than that of the A22 gene and a moderate level was maintained for 49 h of culture (Fig. 6). These transcripts are speculated to have originated from GN for the following reasons. Just after isolation from the anther, no fluorescent signal was observed in either GN or VN of Stage II pollen (Fig. 4g). With the passage of culture time, however, a weak signal appeared in GN but not in VN (Fig. 5e). As described above, similar results were obtained in the expression analysis using the transgenic plant possessing 1600 bp fragment of 5′-upstream of A22 gene instead of A22pro (840 bp) in the transgene. If A22pro and the endogenous promoter of A22 gene are regulated under the same transcriptional control also in Stage II pollen, the difference in the signal intensity for the transcripts between H3sGFP and A22 gene (Fig. 6) may be due to the difference in the half-lives of their transcripts.
Serial observation of pollen undergoing dedifferentiation
Pollen grains freshly isolated from flower buds with a corolla length of 24 mm consisting mainly of Stage III and IV pollen were embedded in medium F with a low gelling-temperature agarose on a chambered cover glass for serial observation under a fluorescent microscope. Just after the pollen was embedded, the fluorescent signal from the reporter protein was observed in most GN and in a small number of VN (2–3%) (Fig. 7e). An intense fluorescent signal was observed in approximately 80% of VN (Fig. 7f) after 30 h of culture and the intensity of the signal was decreased after 75 h (Fig. 7g). The signal in VN disappeared before pollen began the first cell division for embryogenesis. Pollen cell division began asynchronously in the population after about 10 days and developed to embryogenic masses (EM) after 21 days in culture at the frequency of 36% (Fig. 7d). The fluorescent signal was not detectable in EM (Fig. 7h). The pollen that showed a fluorescent signal only in GN died without beginning embryogenic cell division certainly. These serial observations indicated that all the EM originated from the pollen that had shown a fluorescent signal in their VN though the pollen showing the signal in VN did not always develop to EM.
Activation of A22pro in VN is dependent on the pollen developmental stage
The pollen isolated from 15, 20, 24, and 28 mm flower buds were cultured in semisolid medium F for 30 h. The frequency of VN with fluorescence in the viable pollen was 0, 97.0, 77.2, and 34.7%, respectively (Table 1). In the population of the youngest pollen no fluorescent signal was observed in VN and no cell division after 3 weeks. In the other three populations, the frequency of fluorescent VN was roughly proportional to the frequency of EM. The frequency of both fluorescent VN and EM in the population decreased with the progress of the developmental stage of pollen (Table 1). It was possible to trace the original pollen of each EM using pictures of the serial observation (Fig. 7). By tracing the origin of 311 EM, we confirmed that the pollen grains that developed to EM had a fluorescent signal in their VN without exception (Table 1).
Conclusion
We isolated two homologous promoters (A22pro and B27pro) that possess regions approximately 300 bp similar to MAR as a conservative sequence. Using tobacco plants transgenic for a vital reporter gene, H3sGFP, under the transcriptional control of these promoters, we confirmed that these promoters were transcriptionally active in the VN of pollen undergoing embryogenic dedifferentiation while they were inactive in the VN of pollen undergoing maturation and in various somatic tissues containing the early embryo derived from the dedifferentiated pollen. Serial observation revealed that all the EM were originated from the pollen that had activated the promoters in their VN. Interestingly, these promoters were also active in GN within a limited expression level in the pollen undergoing maturation and germination. These promoters seemed to possess plural cis-elements activated in the two different developmental processes, the embryogenic dedifferentiation of vegetative cell and the gametogenesis. Identification of the cis-elements activated in GN and in VN is an interesting issue for future studies.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 17:3389–3402
Bode J, Schlake T, Rios-Ramirez M, Mielke C, Stengert M, Kay V, Klehr-Wirth D (1995) Scaffold/matrix-attached regions: Structural properties creating transcriptionally active loci. Int Rev Cytol 162A:389–454
Bode J, Stengert-Iber M, Kay V, Schlake T, Dietz-Pfeilstetter A (1996) Scaffold/matrix-attached regions: Topological switches with multiple regulatory functions. Crit Rev Eukaryot Gene Expr 6:115–138
Boisnard-Lorig C, Colon-Carmona A, Bauch M, Hodge S, Doerner P, Bancharel E, Dumas C, Haseloff J, Berger F (2001) Dynamic analyses of the expression of the HISTONE::YFP fusion protein in Arabidopsis show that syncytial endosperm is divided in mitotic domains. Plant Cell 13:495–509
Chiu WI, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6:325–330
Dunwell JM, Sunderland N (1974) Pollen ultrastructure in anther cultures of Nicotiana tabacum. I. Early stage of culture. J Exp Bot 25:352–361
Guha S, Maheshwari SC (1964) In vitro production of embryos from anthers of Datura. Nature 204:497
Guha S, Maheshwari SC (1966) Cell division and differentiation of embryos in the pollen grains of Datura in vitro. Nature 212:97–98
Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27:297–300
Horsch RB, Fry JE, Hoffmann NL, Wallroth M, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring cloned genes into plants. Science 227:1229–1231
Jain SM, Sopory SK, Velleux RE (1996) In vitro haploid production in higher plants, vol 1–5. Kluwer Academic, Dordrecht, The Netherlands
Kyo M (1990) Effect of EDTA and acidified medium on the dedifferentiation of immature pollen in a tobacco pollen culture. Plant Cell Physiol 31:1249–1251
Kyo M, Harada H (1986) Control of the developmental pathway of tobacco pollen in vitro. Planta 168:427–432
Kyo M, Miyatake H, Mamezuka K, Amagata K (2000) Cloning of cDNA encoding NtEPc, a marker protein for the embryogenic dedifferentiation of immature tobacco pollen grains cultured in vitro. Plant Cell Physiol 41:129–137
Kyo M, Hattori S, Yamaji N, Pechan P, Fukui H (2003) Cloning and characterization of cDNAs associated with the embryogenic dedifferentiation of tobacco immature pollen grains. Plant Sci 164:1057–1066
Liu YG, Mitsukawa N, Ousumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Michalowski SM, Allen GC, Hall GE Jr, Thompson WF, Spiker S (1999) Characterization of randomly-obtained matrix attachment regions (MARs) from higher plants. Biochemistry 38:12795–12804
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue culture. Physiol Plant 15:473–497
Pechan PM, Smykal P (2001) Androgenesis: affecting the fate of male gametophytogenesis. Physiol Plant 111:1–8
Reynolds TL (1997) Pollen embryogenesis. Plant Mol Biol 33:1–10
Rogers HJ, Bate N, Combe J, Sullivan J, Sweetman J, Swan C, Lonsdale DM, Twell D (2001) Functional analysis of cis-regulatory elements within the promoter of the tobacco late pollen gene g10. Plant Mol Biol 45:577–585
Sakai H, Aoyama T, Oka A (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J 24:703–711
Singh M, Bhalla PL, Xu H, Singh MB (2003) Isolation and characterization of a flowering plant male gametic cell-specific promoter. FEBS Lett 542:47–52
Touraev A, Ilham A, Vicente O, Heberle-Bors E (1996) Stress-induced microspore embryogenesis in tobacco: an optimized system for molecular studies. Plant Cell Rep 15:561–565
Triglia T, Peterson MG, Kemp DJ (1988) A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res 16:8186
Acknowledgements
We thank Drs. Y. Niwa and H. Kobayashi (University of Shizuoka) for supplying the plasmid, pTH-2, harboring the sGFP gene. We thank T. Hitomi for his valuable cooperation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Sato
The nucleotide sequences of A22 cDNA and the 5′-upstream regions of genes corresponding to A22 and B27 reported in this paper have been submitted to DDBJ/EMBL/GenBank under accession numbers AB186042, AB186041, and AB199743, respectively.
Rights and permissions
About this article
Cite this article
Yamaji, N., Kyo, M. Two promoters conferring active gene expression in vegetative nuclei of tobacco immature pollen undergoing embryogenic dedifferentiation . Plant Cell Rep 25, 749–757 (2006). https://doi.org/10.1007/s00299-005-0076-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-005-0076-3