
Abstract
A protocol for the transformation of castor embryo axes using the pCAMBIA vector 1304 in disarmed Agrobacterium tumefaciens strain EHA105 is presented. Co-cultivated explants were initially subjected to expansion and proliferation on MS medium with 0.5 mg l−1 TDZ followed by three cycles of selection on medium with 0.5 mg l−1 BA and increasing concentrations of hygromycin (20–40–60 mg l−1). Selected shoot clusters were transferred to medium with 0.5 mg l−1 BA for proliferation and 0.2 mg l−1 BA for shoot elongation. Elongated shoots were rooted on half-strength MS medium with 2.0 mg l−1 NAA. The presence and stable integration of the hpt gene was confirmed through PCR, RT-PCR, PCR-Southern blot, sequence analysis, Southern blot analysis and PCR analysis of progeny. Southern blot analysis of the primary transformants showed single copy integration and progeny analysis revealed monogenic inheritance of the introduced gene. This paper reports the first successful attempt at producing transgenic castor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Castor (Ricinus communis L.) is an important non-edible oilseed crop widely cultivated in tropical, sub-tropical and temperate countries for its high utilitarian value (Atsmon 1989). Castor oil was primarily used for medicinal purposes and as a general industrial lubricant. Subsequently, castor seed oil and its derivatives have become important commodities and an increasing number of uses are being found for them in the industrial world. The derivatives are used in a range of sectors including agriculture, the textile industry, paper industry, plastics engineering, rubber and pharmaceuticals (Vignolo and Naughton 1991). The use of castor seed as an excellent model system for studying seed development per se and in biochemical and molecular investigations of several metabolic pathways has been reviewed (Molina and Schobert 1995).
Globally, cultivation of castor is constrained by the vulnerability of the improved cultivars to insect attack. Reliable sources of resistance to the major insect pests are rather limited in the available germplasm of this monotypic genus. The other major problem limiting castor cultivation in the industrialized nations is the high level of toxicity of the existing cultivars of castor due to the presence of the protein ricin which poses a serious threat to the health of the growers, seed processors and consumers. Attempts are being made to develop castor with reduced toxicity by combining conventional breeding approaches and transgenic technology (Auld et al. 2001). In Auld et al.’s (2001) studies, traditional breeding methodology led to the generation of F6 lines of castor that have a 70–75% reduction in ricin and Ricinus communis agglutinin toxins, while the transgenic approach is at the stage of evaluation for optimum tissue and developmental timing for the highest efficiency of transformation through Agrobacterium tumefaciens-mediated gene transfer.
Genetic improvement of any crop species through genetic engineering techniques requires an efficient in vitro regeneration system, which is rapid, reproducible and applicable to a broad range of genotypes. However, in the case of castor, research efforts for the last two decades have failed to provide a reliable protocol of in vitro plant regeneration. Reports on in vitro studies in castor suffer from non-reproducibility, low multiplication rates and involvement of pre-existing meristems (Athma and Reddy 1983; Reddy et al. 1987; Sangduen et al. 1987; Reddy and Bahadur 1989; Sarvesh et al. 1992; Molina and Schobert 1995; Sujatha and Reddy 1998).
Genetic engineering has become a necessary tool for the improvement of cultivars of this monotypic genus to confer resistance to biotic stresses and to lower the toxicity of seed meal. Difficulties in tissue culture-related regeneration have compelled researchers to adopt meristem-based transformation methods that have revolutionized plant genetic engineering of major agronomic crops, which were considered recalcitrant to in vitro manipulations (Potrykus 1991). The present investigation has been undertaken to optimize conditions for A. tumefaciens mediated transformation using the meristem proliferation system developed at our laboratory (Sujatha and Reddy 1998).
Materials and methods
Plant material and bacterial strains
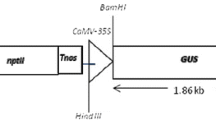
Seeds of castor cv. DCS-9 (Jyoti) were obtained from the Project Coordinating Unit at the Directorate of Oilseeds Research, Hyderabad, India. The disarmed Agrobacterium strain EHA105 harbouring the binary vector pCAMBIA 1304 (cambia@cambia.org.au), which contains a gus gene (uid-A) as the reporter gene and a hpt gene as a selectable marker, both driven by the CaMV 35S promoter, was used for most of the transformation experiments.
Explant preparation and transformation
Mature seeds were decoated and rinsed in running tap water for 30 min. The decoated seeds were surface sterilized with 0.1% (w/v) aqueous mercuric chloride solution for 12 min and subsequently rinsed 5 times with sterile distilled water. The material was blotted dry on sterile filter paper. The endosperm was carefully dissected to expose the embryos. Leaving the papery cotyledons on the endosperm, the embryo axes were excised and implanted on the culture medium poured into 9.0-cm glass Petri dishes. MS (Murashige and Skoog 1962) basal salt medium with 3% sucrose, pH 5.8 and solidified with 0.8% agar (Hi-media, Mumbai) was used in all the experiments. The cultures were maintained at 26±2°C under a 16/8-h photoperiod with light provided by cool white fluorescent lamps at an intensity of 30 μmol m−2 s−1.
Selection of putative transformed shoots
Information with regard to the effective concentration of the commonly used selection agents, viz. hygromycin and kanamycin, is not available for castor. To determine these, the embryo axes and proliferating shoot cultures of non-transformed (control) embryo axes were cultured on MS medium supplemented with 0.5 mg l−1 BA and different concentrations of kanamycin (dilution series 0–500 at 50 mg l−1 increments) and hygromycin (dilution series 0–100 at 5 mg l−1 increments up to 30 mg l−1, and 10 mg l−1 increments thereafter) individually. The antibiotics were filter-sterilized and added to the autoclaved medium. The cultures were scored for survival and shoot proliferation at 3-day intervals up to 30 days.
Transformed shoots are either subjected to selection pressure of three to five cycles at a fixed concentration of the antibiotic or are transferred to selection media with increasing concentrations of the antibiotic. To determine this for castor, three cycles of selection of 15 days’ duration were used. In the first cycle, five concentrations of hygromycin (10–50 mg l−1) were tested. In the second cycle of selection, the concentration of the antibiotic was increased by 20 mg l−1 for each of the tested concentrations and similarly in the third cycle of selection.
The effect of the antibiotic declines under light. However, when meristem-based transformation is employed, light is required for efficient shoot proliferation. The embryo axes were cultured on medium with 0.1 mg l−1 BA for 15 days followed by transfer to the same medium but with an increasing concentration of hygromycin (20–40–60 mg l−1) to get whole plantlets bypassing shoot proliferation. Alternatively, the embryo axes were cultured on medium supplemented with 0.5 mg l−1 TDZ followed by three cycles of selection on medium with 0.5 mg l−1 BA and an increasing concentration of hygromycin (20–40–60 mg l−1) to facilitate meristem proliferation. In these two experiments, the number of healthy proliferating cultures was scored at the end of each selection cycle.
Transformation and plant regeneration
Agrobacterium tumefaciens strain EHA105 (Hood et al. 1993) harbouring pCAMBIA 1304 was grown on YEM (0.4 g l−1 yeast extract, 10 g l−1 mannitol, 0.1 g l−1 NaCl, 0.2 g l−1 MgSO4.7H2O, 0.5 g l−1 K2HPO4, 15 g l−1 agar) medium containing 50 mg l−1 each of kanamycin and rifampicin. A single bacterial colony was inoculated into 50 ml liquid YEM containing the same antibiotics and grown overnight at 28°C on a shaker at 180 r.p.m. Five milliliters of this overnight culture was reinoculated into 50 ml fresh YEM medium containing 50 mg l−1 kanamycin and 10 mg l−1 rifampicin and grown overnight. Bacteria were pelleted at 5,000 r.p.m. for 10 min and resuspended in 25 ml hormone-free liquid MS medium with 3% sucrose and appropriate dilutions were made to give a bacterial concentration of 6.7×107, 1×108, 2×108, 4×108, 8×108 cells/ml. Embryo axes isolated from mature seeds were pre-cultured on medium supplemented with 0.1 mg l−1 BA for 5–7 days prior to infection with Agrobacterium. The seedlings were injured by two strokes of a sharp pointed blade (no. 11) in the meristematic region, which is distinguishable by its characteristic swelling. The radicular regions and cotyledonary extensions were removed from the germinated embryo axes. The processed explants were immersed in bacterial suspension and subjected to vacuum infiltration for 30 min. Subsequently, the explants were blotted dry on sterile filter paper and co-cultivated for 5 days in Petri dishes containing full-strength MS basal salt medium and incubated in the dark at 26±2°C. To enhance the penetration of the Agrobacterium vector into the target tissues, explants were subjected to shaking (180 r.p.m) for 10–30 min during the bacterial incubation; macerated with glass beads; treated with pectinase (0.1–2.0%) or acetosyringone (0–200 μM). To assess the effect of the co-cultivation period on the frequency of transformation, the infected embryo axes were cultured for 2–10 days on the co-cultivation medium.
Following co-cultivation, the explants were washed with 250 mg l−1 cefotaxime for 2 min, rinsed with sterile distilled water 3 times for 5 min each with constant stirring, and blotted dry on sterile filter paper. The procedure optimized for explant preparation, co-cultivation, selection and recovery of whole plantlets is presented in Fig. 1. The rooted shoots were transferred to sterile vermiculite and maintained under high humidity for 10 days. Established plantlets were transferred to soil and grown to maturity. Seeds were collected from the primary and secondary racemes of the T0 plants and the T1 generation was raised.

Procedure for transformation of embryo axes from mature seeds of castor
Analysis of putative transformants
Histochemical assay of Uid A gene expression
The gus histochemical assay was carried out according to Jefferson (1987) with few modifications. Putatively transformed shoots and shoot cultures recovered on hygromycin-selection media were used in the assays. The materials were incubated overnight at 37°C in the substrate solution containing 1 mM X-Gluc in 0.05 M phosphate buffer (pH 7.0) and 30% Triton X-100. Following X-Gluc staining, the shoots were bleached in 95% (v/v) ethanol and viewed under a stereo binocular microscope.
Molecular analysis
DNA extraction and PCR analysis
Total genomic DNA was isolated from young leaves of putative transformants (T0), T1 progeny derived from PCR positive T0 plants and untransformed (control) plants using the CTAB method (Doyle and Doyle 1987). The forward and reverse primers (Bangalore Genei, India) used for the hpt gene were 5′ CAC AAT CCC ACT ATC CTT CGC 3′ and 5′ GCA GTT CGG TTT CAG GCA GGT 3′, respectively, to amplify a 520-bp fragment. The PCR reaction mixture (20 μl) contained 0.48 U Taq DNA polymerase, 10 mM TRIS–HCl (pH 9.0), 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, 150 μM of each dNTP, 1 μl of each forward and reverse primer at a final concentration of 0.25 μM and 100 ng template DNA. For the positive control, 50 pg of the pCAMBIA 1304 DNA was used. DNA from untransformed castor plant was used for the transformation control and reaction mix without DNA as a negative control. The PCR reaction profile included 30 cycles of strand separation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 1 min. The program was extended for 5 min at 72°C. The amplification products were analyzed on 1.4% agarose–ethidium bromide gels. Further, to overrule the possibility of non-specific amplification in PCR, the PCR-amplified fragment generated using primers specific to the hpt gene were sequence characterized (Bioserve, India).
RT-PCR analysis of the putative transformants was carried out using an RT-PCR kit (Bangalore Genei) on the total RNA isolated with the Trizol reagent (Invitrogen) according to the manufacturer’s instructions.
Southern hybridization
Ten micrograms of purified genomic DNA was digested overnight with Xho1 (10 units/μg DNA) in appropriate buffer at 37°C overnight to release a 1,094-bp fragment. The digested DNA was electrophoresed on a 0.8% gel in TRIS–acetate/EDTA buffer and blotted on to a nitrocellulose membrane (Hybond+, Sigma). The 520-bp amplified product of hpt was labelled by random priming using [32P]-dCTP. Hybridization and autoradiography was according to Sambrook et al. (1989).
Results and discussion
Preliminary experiments on the effect of different physical and biochemical variables that can enhance the frequency of transient gus expression were carried out. The variables included bacterial strains (LBA4404 and EHA105 harbouring pCAMBIA 1304 plasmid), bacterial cell density (6.7×107, 1×108, 2×108, 4×108, 8×108 cells/ml), bacterial incubation period (10, 20, 30 min), meristematic explant type (cotyledon node, embryo axes from mature seed, shoot apices of seedling explants and embryos from mature and immature seeds), MS salt strength during co-cultivation (full, 1/2, 1/4, 1/8), genotypes (48-1, Aruna, VP-1, DCS-9, Bhagya), acetosyringone—a phenolic compound which acts as an inducer of vir genes (0–200 μM)—and micro-wounding (glass beads, pricking with hypodermic needle, incision on meristematic zone with blade, particle gun bombardment at 1,100 psi using uncoated microcarriers). The effect of all these parameters/treatments with the exception of explant and bacterial cell density was found to be non-significant. Transient gus expression at a bacterial density of 4 and 8×108 cells/ml was 88.9–97.2% while it was 43–50% at 6.7×107, 1×108, 2×108 cells/ml. As the transformation is meristem based, it was difficult to get rid of the bacterium when used at higher density (4 and 8×108 cells/ml). Hence, all the experiments were carried out with A.tumefaciens strain EHA105 harbouring the pCAMBIA 1304 plasmid, the cv. DCS-9, bacterial density of 2×108 cells/ml with 30-min incubation and co-cultivation on medium with full-strength MS medium.
Access to a prolific system of shoot proliferation from bud explants is a prerequisite for meristem-based transformation. Of the various bud explants, embryo axes from mature seeds were found to be ideal owing to their extensive proliferative ability. Protocols for shoot proliferation from various meristematic explants of castor are available (Athma and Reddy 1983; Reddy et al. 1987; Sangduen et al. 1987; Molina and Schobert 1995; Sujatha and Reddy 1998). Among these, the protocol developed in our laboratory (Sujatha and Reddy 1998) is superior in terms of the enormous proliferative ability of the meristems. In this particular study, differential effects of various cytokinins on shoot proliferation from bud explants was assessed and for the first time we demonstrated the usefulness of TDZ for castor multiplication and also revealed its carryover effect. Taking into account the enormous expanding ability of the meristematic zone of the embryo axes on medium supplemented with TDZ, the present investigation was undertaken to transform castor via A. tumefaciens-mediated gene transfer. TDZ is becoming an integral factor in the genetic improvement of woody species owing to the enhancement of receptivity of the meristematic regions with TDZ pre-treatment prior to transformation (Huetteman and Preece 1993).
Effect of antibiotics and selection regime
The sensitivity of embryo axes and well-developed shoots to kanamycin (0–500 mg l−1) and hygromycin (0–100 mg l−1) was tested. On medium with kanamycin, shoots bleached with increasing concentrations of the antibiotic and 100% bleaching were observed within 4 weeks on 200 mg l−1 while the embryo axes remained apparently healthy even on the highest concentration tested. Explants placed on medium containing hygromycin became necrotic (Fig. 2a) and hence, a selection regime with this antibiotic was devised for castor.

Agrobacterium tumefaciens-mediated transformation of castor. a Selection of putative transformants on hygromycin, b shoot elongation on medium supplemented with 0.5 mg l−1 BA, c transgenic shoot showing GUS expression, d acclimatized transgenic plant
The results of the experiment using increasing concentrations of hygromycin showed a drastic decline in the survival frequency of the explants on medium with 30 mg l−1 and higher concentrations of the antibiotic, and the shoot cultures failed to survive during the second cycle of selection (Table 1). On medium with an initial hygromycin concentration of 10 mg l−1, shoots continued to grow during the third cycle of selection. However, on medium with an initial antibiotic concentration of 20 mg l−1, death was not as sudden as observed with higher concentrations. Thus, an initial concentration of 20 mg l−1 was found optimum, as it was not detrimental to transformed shoots or as slow in its effect as that of the lower concentration, which could lead to the recovery of escapes. To strike a balance between the elimination of untransformed shoots and proliferation of putative transformed shoots, a selection regime of 20–40–60 mg l−1 hygromycin was adopted. In general, an increased concentration of the antibiotic is beneficial over a fixed concentration during different stages of selection as it permits the expression and growth of transformed shoots in the initial stages with a lower concentration of the antibiotic followed by the death of the untransformed shoots with enhanced levels of the antibiotic. Meristem-based transformation leads to the recovery of chimeric shoots. To circumvent this problem, a stringent selection regime involving three cycles of selection with hygromycin (20–40–60 mg l−1) was found to be ideal.
The interaction of hygromycin and light was not significant during the first cycle of selection. The explants survived regardless of the photoperiod and the method of shoot development. However, the effect of hygromycin was found to be significant during the second cycle of selection and it was more pronounced in shoots on medium supplemented with TDZ. Differences due to light on seedling development of embryo axes maintained on medium with 0.1 mg l−1 BA were not significant, while shoot proliferation on medium supplemented with 0.1 mg l−1 TDZ was completely inhibited in the dark. Hence, selection of castor shoots on hygromycin was continued under a 16/8-h photoperiod. Irrespective of the photoperiod, the control explants failed to survive in the third cycle of selection on medium with 60 mg l−1 hygromycin.
Effect of physical treatments
The effect of physical treatments in terms of transient gus expression were found to be significant (Table 2). Regardless of the type of physical injury, a 5-day co-cultivation period resulted in a higher frequency of gus expression as compared to a 3-day co-cultivation period. Pricking of embryo axes with needles followed by evacuation or agitation and blade injury in conjunction with evacuation gave maximum gus expression. Frequency of gus expression in enzyme-treated explants increased with increasing concentration of pectinase but was lower as compared to mechanical wounding.
The effect of enzyme pretreatment on subsequent survival on selection media was assessed (Table 3). Variation due to pectinase treatment of survival of shoot cultures subjected to the first cycle of selection was not profound. However, a drastic decline in shoot survival was observed during the second and third cycles of selection. Enzyme concentrations of 1.0 and 2.0% proved detrimental for meristem proliferation and shoot cultures became completely necrotic on the third selection cycle. Shoot recovery following the three cycles of selection was maximum (7.3%) in embryo axes injured with a blade and subjected to agitation.
Injuring a large number of growing embryo axes individually is not only a tedious process but also results in considerable damage to the surrounding tissue which could lead to a low recovery of transformed cells. Hence, enzyme pre-treatments were tested in order to assess the possibility for scaling-up the co-cultivation process. The beneficial effect of macerating enzymes has been successfully demonstrated in a meristem-based transformation of sunflower, and the enhancement of transformation was attributed to the enlargement of the area where Agrobacterium can attach to the meristematic cells and Vir induction with enzyme treatment (Alibert et al. 1999). In contrast, pectinase treatment of castor embryo axes produced an inhibitory effect on explant development and tissue proliferation. Further investigations are required to improve T-DNA transfer from A. tumefaciens to embryo axes by using lower concentrations of macerating enzymes, antioxidants like cysteine and ascorbic acid or subjecting the explants to sonication as done in soybean (Santarem et al. 1998; Olhoft and Somers 2001).
Co-cultivation period
Differences due to the co-cultivation period were evident during the third selection cycle (Table 4). Co-cultivation of embryo axes for 10 days resulted in a high frequency of transformation as revealed by the high survival frequency. However, shoot proliferation was poor and was accompanied by bacterial overgrowth at all stages of selection and failed to be controlled even on the third cycle of selection. In general, a 2- to 7-day co-cultivation period is considered ideal for Agrobacterium-mediated transformation in many plant species. Hence, in the present investigation a 5-day co-cultivation period was preferred.
Most of the studies on determining factors for enhancing transformation efficiency through A. tumefaciens-mediated gene transfer employ a transient gus assay as an index of T-DNA transfer. However, in the present study inferences were drawn based on the frequency of shoot survival after three cycles of selection. This methodology is promising as the data are based on stable integration of the introduced gene.
Shoot development and plantlet establishment
Embryo axes developed into whole plantlets when cultured on medium supplemented with 0.1 mg l−1 BA or produced multiple shoots with little or no base callusing on medium with 0.5 mg l−1 TDZ. On medium with 0.1 mg l−1 BA for 1 week prior to co-cultivation, the embryo axes elongated and showed characteristic swelling at the meristematic region. Removal of radicular regions followed by co-cultivation and culture of embryo axes on medium with 0.5 mg l−1 TDZ resulted in enormous expansion of the meristematic region with a large number of tiny green-coloured protuberances. Transfer of these cultures to selection medium with 0.5 mg l−1 BA and hygromycin (20–40–60 mg l−1) led to the differentiation of these protuberances into shoot-like structures. During the second and third cycles of selection, non-transformed cultures became necrotic while putative transformants continued shoot proliferation. Transfer of cultures subjected to three cycles of selection to medium with 0.2 mg l−1 BA facilitated shoot elongation (Fig. 2b). Histochemical gus analysis of the shoots recovered after three cycles of selection were positive for gus expression (Fig. 2c). The intensity of gus staining was higher in young leaves and shoot primordia. Castor pollen grains have endogenous gus activity and, hence, cannot be used in an analysis of transgenic plants. All the elongated shoots with two or three distinct nodes developed roots on medium supplemented with 2.0 mg l−1 NAA. The rooted shoots were successfully acclimatized by transferring them to sterile vermiculite and maintaining them under high humidity for 10 days (Fig. 2d).
Molecular analysis
The putative transformants that were obtained after three cycles of selection were PCR positive for the hpt gene and the primers amplified a 520-bp fragment (Fig. 3a). Amplified product was not detected in untransformed shoots (control). The fidelity of the amplified gene fragment was verified by subjecting the PCR gels to Southern blot hybridization (data not shown). The sequence of the amplified fragment was in conformity with the introduced gene sequence. RT-PCR analysis of one of the putative transformants revealed the presence of the 520-bp band (Fig. 3b). Southern blot analysis of genomic DNA showed one copy of the hpt gene (Fig. 3c). Hybridization signals were not detected in the digested DNA of the control plant. Hybridization of PCR-amplified products with the probe showed a signal at the 520-bp position while in the case of genomic DNA digested with Xho1 the DNA fragments hybridized at the 1,094-bp position. The plants that were PCR positive for the hpt gene were advanced to the next generation by selfing the racemes of the T0 plants. PCR analysis of the progeny of three independent transformants using hpt-specific primers showed segregation for the presence and absence of the hpt band in a 3:1 ratio with a χ2 of 0.46 at P=0.5 (Fig. 3d).

Molecular analysis of transgenic plants. a PCR analysis of T0 plants showing amplification of a 520-bp fragment of the hpt gene. Lane M λ DNA double digest with EcoR1/HindIII; lane 1 no DNA control; lane 2 pCAMBIA 1304 DNA; lane 3 DNA from untransformed plant (control); lanes 4, 20 DNA from putative transformants. b RT-PCR of the cDNA showing amplification of a 520-bp fragment of the hpt gene and transcript. Lane M λ DNA double digest with ECoR1/HindIII, lane 2 no DNA control, lane 3 DNA from a putative transformant, lane 4 cDNA from a putative transformant, lane 5 cDNA from untransformed plant (control). c Southern blot analysis of RT-PCR-positive plant using a hpt-amplified fragment as the probe. Lane 1 Undigested pCAMBIA 1304 DNA; lane 2 pCAMBIA 1304 DNA digested with Xho1, lane 3 DNA from RT-PCR positive plant digested with Xho1, lane 4 DNA from untransformed plant (control) digested with Xho1. d PCR analysis of T1 progeny showing amplification of a 520-bp fragment of the hpt gene. Lane M λ DNA double digest with ECoR1/HindIII; lane 1 pCAMBIA 1304 DNA; lane 2 no DNA control; lane 3 DNA from untransformed control; lanes 4,29 DNA from T1 progeny of RT-PCR and Southern blot-positive plant
This study is the first successful attempt to develop a stable transformation system for castor via A. tumefaciens-mediated transfer using embryo axes from mature seeds. With this protocol, a primary transformant can be developed within 5 months from culture initiation. Averaged over several experiments, the frequency of recovery of putative transformants after three cycles of selection worked out at 1 in 1,200 (0.08%) embryo axes cultured. To date, there are no protocols described for the genetic transformation of castor using vegetative explants. The method of Agrobacterium-mediated transformation of castor through vacuum infiltration of wounded flower buds has been patented by McKeon et al. (2003; US patent no. 6,620,986). Until a reliable system of plant regeneration is developed for castor, the meristem-based transformation system described in this report serves as a useful tool for the introduction of desirable genes into this highly industrialized crop.
Abbreviations
- BA:
-
N6-Benzyladenine
- CTAB:
-
Cetyl trimethyl ammonium bromide
- DMRT:
-
Duncan’s multiple range test
- GUS:
-
β-Glucuronidase
- gus :
-
β-Glucuronidase gene
- hpt :
-
Hygromycin phosphotransferase gene
- MS:
-
Murashige and Skoog basal salt media
- NAA:
-
α-Naphthaleneacetic acid
- PCR:
-
Polymerase chain reaction
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- TDZ:
-
1-Phenyl-3-(1,2,3-thiadiazol-5-γl) urea (thidiazuron)
- X-Gluc:
-
5-Bromo 4-chloro 3-indolyl β-D-glucuronide
References
Alibert B, Lucas O, Le Gall V, Kallerhoff J, Alibert G (1999) Pectolytic enzyme treatment of sunflower explants prior to wounding and cocultivation with Agrobacterium tumefaciens, enhances efficiency of transient β-glucuronidase expression. Physiol Plant 106:232–237
Athma P, Reddy TP (1983) Efficiency of callus initiation and direct regeneration from different explants of castor (Ricinus communis L). Curr Sci 52:256–257
Atsmon D (1989) Castor. In: Robbelen G, Downey RK, Ashri A (eds). Oil crops of the world their breeding and utilization. McGraw-Hill, New York, pp 438–447
Auld DL, Rolfe RD, McKeon TA (2001) Development of castor with reduced toxicity. J New Seeds 3:61–69
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgen Res 2:208–218
Huetteman CA, Preece JE (1993) Thidiazuron: a potent cytokinin for woody plant tissue culture. Plant Cell Tiss Org Cult 33:105–119
Jefferson RA (1987) Assaying chimeric genes in plants: the gus fusion system. Plant Mol Biol Rep 5:387–405
Molina SM, Schobert C (1995) Micropropagation of Ricinus communis. J Plant Physiol 147:270–272
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Olhoft PM, Somers DA (2001) l-Cysteine increases Agrobacterium-mediated T-DNA delivery into soybean cotyledonary-node cells. Plant Cell Rep 20:706–711
Potrykus I (1991) Gene transfer to plants: assessment of published approaches and results. Annu Rev Plant Physiol Plant Mol Biol 42:205–225
Reddy KRK, Bahadur B (1989) Adventitious bud formation from leaf cultures of castor (Ricinus communis L.). Curr Sci 58:152–154
Reddy KRK, Rao GP, Bahadur B (1987) In vitro morphogenesis from seedling explants and callus cultures of castor (Ricinus communis L.). Phytomorphology 37:337–340
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Sangduen N, Pongtongkam P, Ratisoontorn P, Jampatas R, Suputtitada S, Khumsub S (1987) Tissue culture and plant regeneration of castor (Ricinus communis L.). SABRAO J 19:144
Santarem ER, Trick HN, Essig JS, Finer JJ (1998) Sonication assisted Agrobacterium mediated transformation of soybean immature cotyledons: optimization of transient expression. Plant Cell Rep 17:752–759
Sarvesh A, Ram Rao DM, Reddy TP (1992) Callus initiation and plantlet regeneration from epicotyl and cotyledonary explants of castor (Ricinus communis L.). Adv Plant Sci 5:124–128
Sujatha M, Reddy TP (1998) Differential cytokinin effects on the stimulation of in vitro shoot proliferation from meristematic explants of castor (Ricinus communis L.). Plant Cell Rep 17:561–566
Vignolo R, Naughton F (1991) Castor: a new sense of direction. Information 2:692–699
Acknowledgements
The authors thank M. Tarakeswari, D. Lakshmi and P. Kanaka Mahalakshmi for their excellent technical help. The financial support from Andhra Pradesh Netherlands Biotechnology Programme is gratefully acknowledged
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R.J. Rose
Rights and permissions
About this article
Cite this article
Sujatha, M., Sailaja, M. Stable genetic transformation of castor (Ricinus communis L.) via Agrobacterium tumefaciens-mediated gene transfer using embryo axes from mature seeds. Plant Cell Rep 23, 803–810 (2005). https://doi.org/10.1007/s00299-004-0898-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0898-4