
Abstract
A reproducible method of Agrobacterium-mediated transformation was developed for Cicer arietinum (chickpea). Initial explants consisted of longitudinal slices from embryonic axes of imbibed, mature seed. The plasmid contained a bi-functional fusion gene conferring both β-glucuronidase and neomycin phosphotransferase activities, under the control of a 35S35SAMV promoter. Using a series of tissue culture media for co-cultivation, shoot initiation and rooting, we recovered transgenic plants from approximately 1.3% of the sliced embryo axes. The addition of a shoot elongation medium to the protocol improved the success rate to 3.1% but increased the time in tissue culture. Inheritance of the gus gene was followed through four generations, both through expression and Southern hybridization assays, and showed the expected Mendelian inheritance pattern.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cicier arietinum L. (chickpea) is a food legume rich in protein suitable for both human and animal consumption. It is a staple in the diet of inhabitants of many of the most populous regions of the world and is grown in over 40 countries. Although India remains the largest producer of chickpeas, production on the Canadian prairies has increased dramatically over the last 5 years.
As with other legume crops, fungal diseases such as Aschochyta blight, in addition to insect pests and nematodes, greatly reduce chickpea yields. As the screening of cultivated genotypes has not identified inherent resistance (Sharma and Ortiz 2000), breeders are turning to wild annual Cicer species as a possible source of desired traits. Unfortunately, interspecific hybridization with chickpea has been largely unsuccessful (Ahmad et al. 1988), and the wild species have not responded well to introgression through conventional breeding techniques for yield improvement (van Rheenen et al. 1993). A reproducible, reliable transformation system, combined with traditional breeding techniques, could aid in improving both the quality and yield of this crop.
Legumes are generally considered recalcitrant to regeneration in tissue culture, thereby hampering genetic transformation. The recovery of transgenic pea (Schroeder et al. 1993; Bean et al. 1997; Grant et al. 1995; Polowick et al. 2000), peanut (Ozias-Akins et al. 1993; Eapen and George 1994; Rohini and Rao 2000; Sharma and Anjaiah 2000) and soybean (Hinchee et al. 1988; Zhang et al. 1999) has been successful. While several reports of in vitro regeneration of chickpea have been published (Murthy et al. 1996; Polisetty et al. 1997; Jayanand et al. 2003), there are few reports of the recovery of transgenic plants (Fontana et al. 1993; Kar et al. 1996; Krishnamurthy et al. 2000).
The goal of the investigation reported here was to develop a reliable transformation method for Cicer arietinum L. We described herein a reproducible method of chickpea transformation, the recovery of transgenic plants and the subsequent testing of both expression and inheritance of the introduced gene.
Materials and methods
Agrobacterium strain and plasmid vector
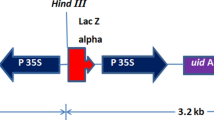
Agrobacterium transformation vector LBG66 (pPBI3008), containing the binary plasmid pPBI3010 was produced as described previously (Polowick et al. 2000). The plasmid contains a bi-functional fusion gene (gus∷nptII) conferring both β-glucuronidase (gus) and neomycin phosphotransferase (nptII) activities (Datla et al. 1991) with a 35S35SAMV promoter (Datla et al. 1993), a NOS (nopaline synthase) terminator and an intron (Vancanneyt et al. 1990). The plasmid was electroporated into the disarmed Agrobacterium strain EHA105 (Hood et al. 1994) to give the vector LBG66.
Plant material and preparation of explants
For the transformation experiments, Cicer arietinum (CDC Yuma; Kabuli-type) seeds were obtained from the University of Saskatchewan’s Crop Development Centre. Batches of seed were surface sterilized for 1 min with 70% ethanol, for 20 min in 20% commercial bleach [Javex; 1% (w/v) NaClO] and then rinsed thoroughly (three times) with sterile distilled water. The seeds (30 per plate) were placed on water agar (0.8%) medium in 150×25-mm plastic petri dishes at 25°C in the dark and imbibed overnight.
Agrobacterium cultures (LBG66) were grown overnight on a rotary shaker at 28°C in the dark in a 2YT medium (Polowick et al. 2000) containing the antibiotics rifampicin (30 mg/l) and gentamycin (25 mg/l). The cultures were centrifuged and resuspended in 2YT without antibiotics to a final concentration of 108 cells per milliliter of culture medium (A660=0.06).
The seed coat and one cotyledon were removed from each imbibed seed and the radicle excised. The remainder of the embryo axis tissue was sliced longitudinally into four or five slices using a surgical blade dipped in the Agrobacterium suspension, as described for pea by Schroeder et al. (1993). The slices were plated on a co-cultivation medium as described below. A 5-μl aliquot of Agrobacterium suspension culture was added to each of the 35–40 slices in a 60×15-mm plastic petri dish. The explants were incubated at 25°C under a 16/8-h (light/dark) photoperiod with light provided by a fluorescent lamp at 35–55 μmol quanta/m2 per second for 4 days and then rinsed in 300 mg/l Timentin on a rotary shaker for a minimum of 1 h. One explant per petri dish was randomly selected and tested for transient gus expression using a X-Gluc assay (described below); the remaining explants were transferred to the medium B5T + 3BA (described below) and returned to the same incubation conditions for 14 days. The surviving explants were transferred to fresh medium every 3 weeks in the series of media described below and illustrated in Fig. 1.

Flowchart of tissue culture media for chickpea transformation and the recovery of transgenic plants. as Acetosyringone (100 μM), B5 B5 salts and vitamins, 3BA benzyladenine (3 mg/l), K kanamycin (concentration in milligrams per liter indicated), MS7 MS medium with B5 vitamins and 1 mg/l BAP, NAA naphthaleneacetic acid (0.18 mg/l), T Timentin (150 mg/l)
Tissue culture media
A number of different media were tested during the course of the study; however, only those which gave consistent positive results are detailed. Except where stated otherwise, all media contained 3% sucrose, were adjusted to a pH of 5.7, solidified with agar (0.8%) and sterilized by autoclaving. All media, except for the co-cultivation medium, contained the commercial antibiotic mixture Timentin (150 mg/l; GlaxoSmithKline, Research Triangle Park, S.C.) to check the growth of residual Agrobacterium. Acetosyringone, kanamycin and Timentin were filter-sterilized prior to incorporation into the culture media.
The co-cultivation medium contained B5 basal salts and vitamins (Gamborg et al. 1968) and 100 μM acetosyringone in 60×15-mm petri dishes.
The shoot induction medium, B5T + 3BA, contained B5 salts and vitamins, 3 mg/l/13.3 μM benzylaminopurine (BAP) and 50 mg/l kanamycin. The first culture cycle (2 weeks) on this medium was in small (60×15-mm) petri dishes; subsequent cycles on this and subsequent media were for 3-week periods and in larger (100×25-mm) petri dishes.
The shoot elongation medium, MS7T (Polowick et al. 2000), consisted of MS medium (Murashige and Skoog 1962) containing B5 vitamins and 1 mg/l/4.4 μM BAP. The kanamycin concentration remained at 50 mg/l for the initial transfer into this medium but was increased to 75 mg/l for the two subsequent cycles.
The rooting medium consisted of B5 basal salts and vitamins, 0.18 mg/l/1.0 μM naphthaleneacetic acid (NAA) with the kanamycin concentration increased to 150 mg/l. Shoots with short roots at the time of transfer were temporarily transferred to Magenta vessels containing B5 basal salts and vitamins and 0.7% agar, but no selection chemical, until the root system was well established (1–3 weeks). Those with roots longer than 3 cm at a time of transfer from the rooting medium were transferred directly into soil.
Rooted shoots were transferred to plastic pots (150×180 mm) containing a commercial soil mixture (Sunshine No. 4, Sun Gro Horticulture, Bellevue, Wash.) and moved to a controlled environment chamber maintained at 20/15°C (day/night) and a 16/8-h (light/dark) photoperiod with light supplied by mixed fluorescent-incandescent lamps at an intensity of 200 μmol quanta/m2 per second . Alternately, the rooted shoots were planted in the same soil mixture in larger [13.6 l; 25-cm (dia)×23-cm (high)] pots and grown under greenhouse conditions with supplementary lighting provided by sodium vapor bulbs to provide a 16/8-h (light/dark) photoperiod.
The young plants were covered with inverted beakers to maintain conditions of high humidity for the first 3–4 days. The beakers were then placed at an angle for an additional 2–3 days to slowly acclimate the plants to the greenhouse atmosphere. The plants were watered with a quarter-strength Hoagland’s nutrient solution until they were well established. After approximately 2 weeks in soil, Nutricote Type 100 (14-14-14) slow-release fertilizer was added to each pot. Wire cages were used to physically support the shoots. Under greenhouse conditions, watering was initially from below, but later watering from both above and below after the plants was established.
Histochemical assays
For visual identification of gus activity in explants at the end of co-cultivation, or in regenerated plants and their offspring, entire explants or individual leaflets were incubated at 37°C for 48 h in a 5-bromo-4-chloro-3-indolyl β-d-glucuronide (X-Gluc) solution (Jefferson 1987). For a quantitative determination of GUS activity, assays using 4-methyl umbelliferyl β-d-glucuronide (MUG; Jefferson 1987) were performed, as previously described (Polowick et al. 2000), on individual leaflets from the regenerated chickpea plants and their offspring. In cases where the X-Gluc histochemical assays indicated a strong reaction, a dilution of the extract was used to avoid depletion of the MUG substrate. Aliquots of the sample were also removed for measurement of the protein concentration using a dye-binding assay (Bradford 1976; BioRad, Hercules, Calif.). The concentration of 4-methyl umbelliferone (MU) was determined with a Perkin-Elmer LS50 fluorometer (PE Applied Biosystems, Foster City, Calif.). GUS activity was expressed as picomoles MU per milligram protein per minute.
Southern hybridization analysis
The presence of the gus gene was confirmed by Southern blot analysis. A CTAB method, modified from Murray and Thompson (1980), was used to extract genomic DNA from four leaflets of the putative transformants and their offspring. DNA was digested with HindIII and probed with an 1,800-bp BamHI/SstI fragment of the plasmid pBI121 (Jefferson 1987), encompassing the entire gus gene, including the intron. The single HindIII site in the plasmid is situated in the promoter, and the expected length of any band hybridizing with the probe would be greater than 4,564 bp. The probe was labeled with [32P]-dCTP using random primer-labeling (GibcoBRL, Gaithersburg, Md.) and hybridized with the QuikHyb system (Stratagene, La Jolla, Calif.).
Segregation analysis
In order to examine the inheritance of the gus gene and the stability of the transformants, we planted the T1, T2 and T3 progeny of the original (T0) transformants under the same conditions as the T0 plants. Leaflets were collected for analysis by both the X-Gluc and MUG assays. In addition, the offspring of 12 different events were subjected to Southern hybridization analysis.
Results and discussion
Transgenic chickpea plants were recovered using several different media pathways. Only those that provided a reasonable frequency and consistency of recovery will be described in detail. Over the 4-day co-cultivation period, the explants (Fig. 2a) enlarged and turned green (Fig. 2b). Although considered necessary in other chickpea studies (Kar et al. 1996), the co-cultivation medium used in the present study (B5) did not contain any growth regulators. When growth regulators (auxin and cytokinin) were added, as described for pea transformation (Polowick et al. 2000), masses of transformed chickpea callus, but no shoots, were produced. Acetosyringone (100 μM) was utilized, based on preliminary studies of transient X-Gluc staining at the end of co-cultivation; in the presence of acetosyringone, there was extensive blue colouring in the explants (Fig. 2c), while in the absence of acetosyringone X-Gluc staining was comparatively minimal (not shown). No transgenic plants were recovered after co-cultivation without acetosyringone, albeit from a small number of explants. Also, results from previous (unpublished) studies on other legumes (e.g. pea and lentil) on the activation of vir genes emphasize the importance of both the presence of acetosyringone and wounding of the explants and support a 4-day co-cultivation period. Acetosyringone was not used in Agrobacterium-mediated chickpea transformation reported by Fontana et al. (1993), Kar et al. (1996) and Krishnamurthy et al. (2000).

Stages of tissue culture for transgenic chickpea plant recovery and GUS (X-Gluc) assays of potential transgenic material. a Explant at the time of co-cultivation. Bar: 1 mm. b Explant at the end of 4 days of co-cultivation. Bar: 1 mm. c X-Gluc assay of explant at the end of co-cultivation. Bar: 1 mm. d Explants on shoot elongation medium. Bar: 10 mm. e Shoot material, all from a single explant, on rooting medium. Bar: 10 mm. f Rooted shoot prior to placement on soil. Bar: 5 mm. g X-Gluc assay on one leaf from a transgenic chickpea plant. Bar: 10 mm
Media previously used for the transformation of slices of pea embryonic axes (P1 and P2; Schroeder et al. 1993) were tested, but the frequency of recovery of transgenic plants was less than 0.5%. Over the course of the study we determined that three transfers through the shoot induction medium (B5T + 3BA; Fig. 2d) were sufficient for shoot induction prior to the transfer to shoot elongation or rooting media.
When the shoot elongation medium (MS7T) was utilized (Fig. 2e), the first cycle was with 50 mg/l kanamycin. For the following two cycles, the kanamycin concentration was increased to 150 mg/l to reduce the possibility of escapes. The same medium was used for shoot elongation in the pea transformation (Polowick et al. 2000).
Shoot proliferation continued on the root induction medium such that, after each passage through the medium, small clumps of shoots could be separated into individual shoots, which in turn produced a small cluster of new shoots at the base. Due to this proliferation, the tissue from one individual slice could spread out to occupy more than one 100-mm petri dish (Fig. 2f); this provided an early suggestion of successful transformation. This continuous subdivision, with the selection of only the most vigorous shoots, possibly reduced the probability of chimaeras. No chimaeras were identified during the course of this study, either from multiple sampling of individuals or from deviations from expected inheritance patterns (see below).
Roots appeared to develop best on shoots that had been separated by breaking the clumps apart at natural points of weakness rather than from those with cut surfaces. Roots were initiated after as few as one to as many as six cycles through the rooting medium. The frequency of rooting was variable (10–60%). However, the large number of shoots produced from transformed explants partially compensated for this; where a large number of shoots were produced from one explant it was not necessary for all of them to root. Generally, no more than 20–25 shoots were maintained from each explant, and there was insufficient space to plant out more than ten plants from each event. In other studies with chickpea, the rooting medium lacked a selection chemical (Fontana et al. 1993; Kar et al. 1996), or potential transgenic shoots were grafted onto etiolated seedlings (Krishnamurthy et al. 2000). Polowick et al. (2000), in work with peas, emphasized the importance of retaining, or even increasing, the concentration of selection chemicals to minimize the number of non-transformed plants that escape selection. In the present study, the concentration of the selection chemical kanamycin was increased to 150 mg/l—double that of the shoot elongation medium.
The transfer of rooted shoots to Magenta jars containing MS medium without growth regulators or kanamycin was intended to encourage both root and shoot growth prior to transplantation into soil. In many cases, the roots developed quickly, elongating more than 3 cm in 1 week (Fig. 2g) and were too long to permit this transfer without causing damage.
When we followed the media pathway leading directly to the rooting medium (right side of Fig. 1), the frequency of recovery of rooted transgenic plants was 1.3% (Table 1), with a mean tissue culture phase, from co-cultivation to planting in soil, of 160 days (range: 127–287 days). The addition of three passages through the shoot elongation medium (MS7T; one with 50 mg/l kanamycin and two with 75 mg/l) to the media pathway increased the frequency of plant recovery to an average of 3.1% but also increased the duration of the tissue culture phase to a mean of 217 days (range: 133−384 days). The activity of the gus gene did not appear to be restricted by this increased period of tissue culture as the mean GUS activity for these plants was greater although much more variable (Table 2). The frequency of recovery reported in other studies ranges from less than 0.4% (Krishnamurthy et al. 2000) to less than 2% (Kar et al. 1996). Fontana et al. (1993) claimed a 4% success rate; however, only three transgenic plants were recovered.
As reported by Krishnamurthy et al. (2000) and also cited in a meeting report (Jaiwal et al. 2001), the recovered plants did not grow well and often did not survive or set viable seed. This was also true of control (non-transgenic) plants and was, therefore, blamed on poor growing conditions. The plants were very sensitive, and it would only take one incident over watering to decimate an entire batch of plants whether from rooted (putative) transgenics or normal seedlings. This made any calculation of survival rates difficult. A variety of soil mixtures, pot sizes, lighting conditions and watering regimes were tested in order to improve survival. Plants grown in larger (13.6 l) pots and under greenhouse conditions grew more vigorously and set more seed than plants grown under controlled growth conditions.
Southern hybridization analysis (Fig. 3a) was only performed on rooted shoots and indicated the number of inserts in each event (Table 2). It also confirmed the expectation that all rooted shoots arising from the same slice/explant are clones. From 56 independent events identified and tested, only two of the recovered (T0) plants were identified as not having a gus gene. In both cases, only one shoot rooted from that event. This indicated that the concentration of selection chemical, especially in the rooting medium, was sufficient to minimize escapes.

Southern hybridization blots of DNA from putative transgenic chickpea plants and their offspring. The DNA was digested with EcoRI and probed with an 1,800-bp BamHI/SstI fragment of the plasmid pBI121. a DNA from putative transformants recovered after co-cultivation with Agrobacterium transformation vector LBG66 (pPBI3010) and selection on kanamycin, b DNA from the original D49 transformant (T 0 ) plus ten of the T1 offspring
The inheritance of the inserted gus gene was assessed by X-Gluc and MUG assays as well as by Southern hybridization analysis. For example, eight of the ten offspring tested from D49 had the same banding pattern on the Southern hybridization blot as the parent (T0) plant (Fig. 3b). This is close to the 7.5 expected with a 3:1 ratio for a single gene insertion (χ2=0.72). Similar patterns were observed with the T1 offspring from other plants (Table 3). In addition, GUS activity was retained in the next generation (Table 3). In one exceptional case, the inserted genes were absent from all ten tested offspring of D52. As this could not be determined while the parent plant was still alive, it cannot be ascertained if this loss was due to poor integration of the gene(s) in the parent plant because of a chimera or due to mislabeling. Inheritance of the inserted genes was not demonstrated in most of previous chickpea studies (Fontana et al. 1993; Kar et al. 1996). Krishamurthy et al. (2000) found that while the nptII gene was detectable by means of PCR analysis, there was no GUS activity in the four viable offspring produced.
Three independent lines (D45, D49, P30) were followed through the T2 generation. On the basis of their MUG assay values (Table 3), the gus gene was inherited in all three lines and its activity did not diminish. In addition, inheritance of the gus gene was followed to the T3 generation of two of the lines (D49, P30) and GUS activity was observed to continue. In the case of P30, the line arising from the single initial T1 seed (Table 3) was determined to be homozygous for the introduced gus gene, as the two bands observed in the parent (T0) plant were found in all 20 of the plants tested from the T3 generation (not shown). In addition, GUS activity was maintained (Table 3), indicating continued inheritance and expression of the gus gene.
The method of chickpea transformation described herein is laborious in the initial preparation of the explants. However, the use of mature seed is advantageous, as it does not require a continuous supply of developing material. In order to be considered truly viable and effective, the method of transformation should be reproducible with a broad range of chickpea genotypes. The cultivar used in this study (CDC Yuma) was selected because it is locally grown rather than because it has any pre-determined responses in tissue culture. Therefore, success with other cultivars should be anticipated. Experiments are in progress with the Kabuli-type CDC Xena. Similarly, cvs. CDC Desiray and Myles are being tested to see if Desi-type seeds have different media requirements. While it is too soon to determine the frequency of success, transgenic plants have been recovered from each of these cultivars, indicating that the method is genotype-independent.
References
Ahmad F, Slinkard AE, Scoles GJ (1988) Investigations into the barrier(s) to interspecific hybridization between Cicer arietinum L. and eight other annual Cicer species. Plant Breed 100:193–198
Bean SJ, Gooding PS, Mullineaux PM, Davies DR (1997) A simple system for pea transformation. Plant Cell Rep 16:513–519
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Datla RSS, Hammerlindl JK, Pelcher LE, Crosby WL, Selvaraj G (1991) A bifunctional fusion between β-glucuronidase and neomycin phosphotransferase: a broad-spectrum marker enzyme for plants. Gene 101:2139–2246
Datla RSS, Bekkaoui F, Hammerlindl JK, Pilat G, Dunstan DI, Crosby WL (1993) Improved high-level constitutive foreign gene expression in plants using an AMV RNA4 untranslated leader sequence. Plant Sci 94:139–149
Eapen S, George L (1994) Agrobacterium tumefaciens-mediated gene transfer in peanut (Arachis hypogaea L.). Plant Cell Rep 13:582–586
Fontana GS, Santini L, Caretto S, Frugis G, Mariotti D (1993) Genetic transformation in the grain legume Cicer arietinum L. (chickpea). Plant Cell Rep 12:194–198
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Grant JE, Cooper PA, McAra AE, Frew TJ (1995) Transformation of peas (Pisum sativum L.) using immature cotyledons. Plant Cell Rep 15:254–258
Hinchee MAW, Connor-Ward DV, Newell CA, McDonnell RE, Sato SJ, Gasser CS, Fischhoff DA, Re DB, Fraley RT, Horsch RB (1988) Production of transgenic soybean plants using Agrobacterium-mediated DNA transfer. Biotechnology 6:915–921
Hood EE, Glevin SB, Melchers LS, Heokema A (1994) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218
Jaiwal PK, Sonia, Upadhyaya KC (2001) Chickpea regeneration and transformation (meeting report). Curr Sci 80:1368–1369
Jayanand B, Sudarsanam G, Sharma KK (2003) An efficient protocol for the regeneration of whole plants of chickpea (Cicer arietinumL.) by using axillary meristem explants derived from in vitro-germinated seedlings. In Vitro Cell Dev Biol Plant 39:171–179
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Kar S, Johnson TM, Nayak P, Sen SK (1996) Efficient transgenic plant regeneration through Agrobacterium-mediated transformation of chickpea (Cicer arietinum L.). Plant Cell Rep 16:32–37
Krishnamurthy KV, Suhasini K, Sagare AP, Meixner M, de Kathen A, Pickardt T Schieder O (2000) Agrobacterium mediated transformation of chickpea (Cicer arietinum L.) embryo axes. Plant Cell Rep 19:235–240
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:473–497
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Murthy BNS, Victor J, Singh RP, Fletcher RA, Saxena PK (1996) In vitro regeneration of chickpea (Cicer arietinum L.): stimulation of direct organogenesis and somatic embryogenesis by thidiazuron. Plant Growth Regul 19:233–240
Ozias-Akins P, Schnall JA, Anderson WF, Singsit C, Clemente TE, Adang MJ, Weissinger AK (1993) Regeneration of transgenic peanut plants from stably transformed embryogenic callus. Plant Sci 93:185–194
Polisetty R, Paul V, Deveshwar JJ, Khetarpal S, Suresh K, Chandra R (1997) Multiple shoot induction by benzyladenine and complete plant regeneration from seed explants of chickpea (Cicer arietinum L.). Plant Cell Rep 16:565–571
Polowick PL, Quandt J, Mahon JD (2000) The ability of pea transformation technology to transfer genes into peas adapted to western Canadian growing conditions. Plant Sci 153:161–170
van Rheenen HA, Pundir RPS, Miranda JH (1993) How to accelerate the genetic improvement of a recalcitrant crop species such as chickpea. Curr Sci 65:414–417
Rohini VK, Rao KS (2000) Transformation of peanut (Arachis hypogaea L.): a non-tissue culture based approach for generating transgenic plants. Plant Sci 150:41–49
Schroeder HE, Schotz AH, Wardley-Richardson T, Spencer D, Higgins TJV (1993) Transformation and regeneration of two cultivars of pea (Pisum sativum L.). Plant Physiol 101:751–757
Sharma KK, Anjaiah V (2000) An efficient method for the production of transgenic plants of peanut (Arachis hypogaea L.) through Agrobacterium tumefaciens-mediated genetic transformation. Plant Sci 159:7–19
Sharma KK, Ortiz R (2000) Program for the application of genetic transformation for crop improvement in the semi-arid tropics. In Vitro Cell Dev Biol Plant 36:83–92
Vancanneyt G, Schmidt R, O’Connor-Sanchez A, Willmitzer L, Rocha-Sosa M (1990) Construction of an intron-containing marker gene: splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium-mediated plant transformation. Mol Gen Genet 220:245–250
Zhang Z, Xing A, Staswick P, Clemente TE (1999) The use of glufosinate as a selective agent in Agrobacterium-mediated transformation of soybean. Plant Cell Tissue Organ Cult 56:37–46
Acknowledgements
The authors gratefully acknowledge the work of Mr. Terry Bethune during the initial stages of the study and the molecular analysis of plants by Ms. Maureen Anderson. The Saskatchewan Pulse Growers Association provided partial funding for this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M.C. Jordan
NRCC Grant No. 46589.
Rights and permissions
About this article
Cite this article
Polowick, P.L., Baliski, D.S. & Mahon, J.D. Agrobacterium tumefaciens-mediated transformation of chickpea (Cicer arietinum L.): gene integration, expression and inheritance. Plant Cell Rep 23, 485–491 (2004). https://doi.org/10.1007/s00299-004-0857-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0857-0