
Abstract
A transformation system for selected mature cork oak (Quercus suber L.) trees using Agrobacterium tumefaciens has been established. Embryos obtained from recurrent proliferating embryogenic masses were inoculated with A. tumefaciens strains EHA105, LBA4404 or AGL1 harbouring the plasmid pBINUbiGUSint [carrying the neomycin phosphotransferase II (nptII) and β-glucuronidase (uidA) genes]. The highest transformation efficiency (4%) was obtained when freshly isolated explants were inoculated with A. tumefaciens strain AGL1. Evidence of stable transgene integration was obtained by PCR for the nptII and uidA genes, Southern blotting and expression of the uidA gene. The transgenic embryos were germinated and successfully transferred to soil.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most characteristic tree species of the Mediterranean ecosystem is the cork oak, Quercus suber L., which in addition to being of great ecological value produces cork, a natural renewable product of economic interest. This species is been actively planted within the framework of the European Union-funded programs of reforestation of abandoned former agricultural lands.
Conventional breeding of cork oak is constrained by its long reproductive cycle, which includes long juvenile periods, and by its complex reproductive characteristics, including self-incompatibility and a high degree of heterozygosis. The cork oak, like other tree species, has undergone relatively little domestication; consequently biotechnology could potentially have a greater impact on forestry and forest products than it has had on agronomic crops. Biotechnological approaches for the improvement of Quercus species, such as in vitro vegetative propagation based on somatic embryogenesis, have been reviewed by Wilhelm (2000). Genetic transformation offers an attractive alternative to breeding because it provides the potential to transfer specific traits into selected genotypes without affecting their desirable genetic background. This is of particular importance in woody species, in which many adaptative and economic traits are under non-additive genetic control and, consequently, a specific genetic make-up has to be transferred to the offspring. Within the family Fagaceae, only Castanea sativa (Seabra and Pais 1997, 1999) and Quercus robur (Roest et al. 1991; Wilhelm et al. 1996) have been transformed. Transgenic studies aimed at shortening the juvenile phase, phytoremediation purposes, alterations of the lignin biosynthesis pathway and increased cellulose accumulation have been carried out (for review, Merkle and Dean 2000; Peña and Séguin 2001). However, several features inherent to the biology of forest species (e.g. the large genome sizes and the recalcitrance of tissues from mature trees to in vitro manipulation) still pose a challenge to researchers. An effective selection can only be performed on mature trees (Zobel and Talbert 1984) because desired traits are commonly expressed only at maturity. These facts, together with the lack of culture systems for the regeneration of plants from selected tree clones, have delayed the development of useful methods for rare woody perennials. Cork oak plantlets can be obtained from embryogenic lines initiated from zygotic embryos (Bueno et al. 1992; Manzanera et al. 1993). Fortunately, repetitive embryogenic lines can also be induced in leaves from seedlings (Fernández-Guijarro et al. 1994, 1995) or even from mature trees of this species (Hernández et al. 2001, 2003b), thereby opening the possibility to manipulate and clone desired genotypes. In fact, plants have been regenerated from several selected trees (Hernández et al. 2003a).
The objective of the investigation reported here was to determine whether a repetitive embryogenic system could be used for Agrobacterium tumefaciens-mediated transformation of selected mature cork oak plants. This study is the first report on the development of a protocol for obtaining transgenic plants from mature selected cork oak trees.
Materials and methods
Plant material and culture conditions
An embryogenic line derived from a selected, more than 100-year-old cork oak tree growing in Torrelaguna (Madrid, Spain) was initiated following the protocol of Hernández et al. (2001). Expanding leaves were used as initial explants. These were taken from epicormic shoots that were forced to sprout from pieces of branches collected from the crown of the tree. Following asepsia and a preconditioning period, the induction protocol consisted of three steps on an expression-proliferation basal medium (MSSH). The first step was accomplished by culture in the presence of 10 μM BA and 10 μM NAA in darkness for 30 days. During the second step the levels of both BA and NAA were reduced to 0.5 μM, and cultures were transferred to a 16/8-h (light/dark) photoperiod at 25±1°C for 30 days. Somatic embryos developed when the leaves were transferred onto the same medium lacking growth regulators under the growing conditions of the second step.
The embryogenic line designated as M10 was used in the transformation studies. It was multiplied by recurrent embryogenesis on MSSH without plant growth regulators (Fernández-Guijarro et al. 1995), which was routinely renewed every 30 days, and maintained at 25±1°C under a 16/8-h (light/dark) photoperiod with light provided by cool-white fluorescent tubes (mixed Sylvania Gro-Lux and Philips) at an intensity of 100 μmol m−2 s−1). Somatic embryos which matured spontaneously were picked out from actively growing cultures, transferred to fresh basal medium and stored in the cold (4±1°C) in the dark for 60 days. For germination, the embryos were moved to a growth chamber under the same conditions used for multiplication. After 30 days, germinating embryos were transplanted to forest containers filled with substrate (Hernández et al. 2003a). Baby food jars capped with Magenta caps (Sigma, St. Louis, Mo.) and sealed with plastic film were used to culture the embryogenic lines as well as for cold treatment and germination.
The effects of the aminoglycoside antibiotics kanamycin (50, 100, 150, 200, 250 mg l−1), G418 (10, 15, 20, 30, 40 mg l−1) and paromomycin (10, 15, 20, 30, 40 mg l−1) were assayed in the MSSH medium before the transformation studies. These antibiotics were obtained from Duchefa (The Netherlands).
Bacterial strains
Three disarmed Agrobacterium tumefaciens strains were used in the transformation experiments: EHA105 (Hood et al. 1990), LBA4404 (Hoekema et al. 1983) and AGL1 (Lazo et al. 1991). These strains harboured the binary vector pBINUbiGUSint (Fig. 1; Humara et al. 1999), a derivative of pBIN19 (Bevan 1984), carrying the neomycin phosphotransferase II (nptII) and the β-glucuronidase (uidA) genes driven by the nopaline synthase promoter (NOS-P) and the promoter of the ubi1 gene of maize polyubiquitin (Christensen and Quail 1992), respectively. Both genes carried the NOS-pA terminator. The uidA gene used in this investigation had the PIV2 intron of the ST-L1 gene from Solanum tuberosum within its coding sequence (uidA-int), thereby preventing its expression in Agrobacterium (Ecker et al. 1986; Vancanneyt et al. 1990).

Structure and restriction map of the T-DNA of Agrobacterium tumefaciens binary vector pBINUbiGUSint (16.1 kb). Ubi1 maize polyubiquitin gene promoter; uidA-int uidA gene with the potato PIV2 intron, nptII neomycin phosphotransferase II gene, NOS-pA polyadenylation signal from the nopaline synthase gene, RB, LB right and left borders of the T-DNA, respectively. Restriction sites for HindIII and BglII are indicated
Bacterial culture and explant inoculation
Bacterial strains were grown at 28°C for 24 h in liquid YEP medium (An et al. 1988) containing 50 mg l−1 kanamycin (Duchefa) at 250–300 rpm. The bacterial suspension was then centrifuged at 4,000 rpm, washed in 10 mM MgSO4 and resuspended in liquid MSSH medium at a bacterial concentration of 0.5 absorbance units (O.D.600 nm).
Proliferating embryonic clusters and isolated embryos were selected and placed in liquid MSSH medium to prevent desiccation while they were collected (Fig. 2a). A small wound was made at the embryo axis while picking them out with the forceps. Explants were incubated in the bacterial solution for 20 min at 25±1°C with gentle shaking. Following inoculation, the embryos were blotted on sterile filter paper and transferred to solid MSSH medium. Co-cultivation was carried out in the dark at 25±1°C for 2 days. The inoculated embryos were washed for 2 h in a liquid MSSH medium containing 600 mg l−1 cefotaxime (Duchefa), blotted on sterile filter paper, transferred to selection medium (MSSH containing 500 mg l−1 cefotaxime and 100 mg l−1 kanamycin) and cultured under a 16/8-h (light/dark) photoperiod (100 μmol m2 s−1) at 25°C. Cultures were transferred weekly to fresh selection medium during the first month and at 2-week intervals thereafter. About 2 months after transformation, white proliferating masses (Fig. 2b) could be detected, and these were transferred to fresh selection medium. After 3 months in the selection medium, those masses showing secondary embryogenesis were transferred and subcultured on proliferation medium without kanamycin. Transgenic clones were then maturated and germinated on medium without antibiotics, as described by Hernández et al. (2003b).

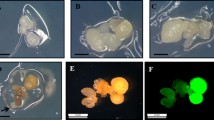
Stages of the cork oak (Quercus suber L.) transformation process. a Somatic embryo cluster used for cocultivation. Bar: 1 mm. b Putative transformed embryo masses surrounded by non-transformed necrotic tissue. c One-month-old transgenic plant grew in vitro. Bar: 1 cm. d Two-month-old transgenic plant growing in Perlite substrate. Bar:1 cm
Molecular analysis of putative transformants
Molecular analysis was performed on putative transgenic and control plants using the PCR and Southern blot. Genomic DNA was extracted from embryos that had proliferated for more than 4 months in selection medium and from non-transformed control explants using the DNeasy® Plant Mini kit for DNA isolation from plant tissue (Qiagen, Germany). The PCR analysis was performed in a Perkin Elmer Gene Amp PCR system 9700 thermocycler (Foster City, Calif.) with 25-μl aliquots of reaction mixture containing 100 ng DNA, 250 μM of each dNTP, 10 pmol of each primer, 1.25 U Bioline Taq DNA polymerase, 2 mM MgCl2 and the 1× corresponding Bioline Taq buffer. The putative transgenic clones were PCR tested for the nptII, uidA and virG genes. Primers used to amplify the 700-bp sequence of the nptII gene were 5′-GAGGCTATTCGGCTATGACTG-3′ (201–222 bp) and 5′-ATCGGGAGCGGCGATACCGTA-3′ (879–900 bp). Primers used to amplify the uidA gene were 5′-GGTGGGAAAGCGCGTTACAAG-3′ (400–420 bp) and 5′-GTTTACGCGTTGCTTCCGCCA-3′ (1,579–1,599 bp). Primers used to amplify the virG gene sequence were forward 5′-AAGGTGAGCCGTTGAAACAC-3′ and reverse 5′-ATCTCAAGCCCATCTTCACG-3′.
Samples were heated at 95°C for 4 min, and then subjected to 35 cycles of 45 s at 95°C, 45 s at 59°C and 1 min at 72°C, followed by an extension cycle at 72°C for 10 min. Conditions for virG were 95°C for 4 min and 35 cycles of 45 s at 95°C, 45 s at 56°C and 1 min at 72°C, followed by an extension at 72°C for 10 min.
For Southern blot analysis, 10 μg DNA from each sample were individually digested with 300 U and 150 U of HindIII or BglII, respectively, for 16 h at 37°C and subsequently separated on a 0.7% agarose gel at 60 V for 5 h. DNA was transferred onto a positively charged nylon membrane (Hybond-N+; Amersham Pharmacia Biotech, Piscataway, N.J.). A HindIII (4.4 kbp) and a HindIII-BglII (3.3 kbp) fragment containing the uidA gene sequence were used as probes following [32P]-labelling by random priming (Rediprim; Amersham, UK). Hybridisation and detection were carried out according to Sambrook et al. (1989).
Histochemical (GUS) analysis of transformants
Embryos that were proliferating in selection medium were also assayed histochemically for GUS activity following a protocol modified from Jefferson (1987). Embryos not exceeding 2 mm in diameter were incubated in the dark for 14 h at 37°C in a solution of 2 mM 5-bromo-4-chloro-3-indolylglucuronide (X-Gluc), 0.5 mM K3Fe(CN)6, 0.5 mM K4Fe(CN)6, 0.01 M Na2 EDTA, 0.1% triton X-100 and 0.1 M phosphate buffer pH 7.0. A low vacuum was applied to ease the inflow of the chemical into the tissues.
Once incubation was carried out, embryos were washed in phosphate buffer and analysed for positive blue colouring.
Statistical analysis
Each experiment was performed three times with a minimum of 150 explants per treatment. Proliferation was measured as the number of embryos that maintained a white-yellowish colour, that appeared to proliferate and that did not become brown with respect to the total number of embryos. These qualitative data were presented as the mean ± standard error and analysed statistically using the chi-square test. All statistical analyses were performed at the 5% level using spss statistical software.
Results and discussion
Various factors influencing the efficiency of T-DNA delivery into cork oak embryogenic cells via A. tumefaciens were evaluated. These factors included the tolerance of the cork oak embryos to cefotaxime, which was used to inhibit Agrobacterium growth following infection (Nauerby and Billing 1997; Humara and Ordás 1999) and to some of the selective agents available for use with the nptII gene.
The cephalosporin cefotaxime was successfully used to suppress the growth of the three Agrobacterium strains tested and was essentially non-toxic to control cork oak embryos (data not shown). However, the proliferation rate of inoculated embryogenic cultures was affected by the bacterial strains tested in the presence of this antibiotic at the beginning of culture. Although there was no early bacterial regrowth after co-cultivation, the infection reduced the proliferation rate of cork oak embryos. Secondary embryogenesis was observed in all control and inoculated explants after 90 days of culture in the presence of cefotaxime.
To develop a selective growth system for genetically transformed cork oak embryos, we studied the effects of kanamycin, G418 and paromomycin at various concentrations. Recurrent embryogenesis was inhibited at all of the concentrations of G418 and paromomycin tested, which led to a high phenol exudation within 30 days (data not shown). In contrast, the presence of 100 mg l−1 kanamycin did not induce phenol exudation and inhibited the process of recurrent embryogenesis (Fig. 3). The frequent (2-week intervals) subculture of the embryos in the presence of 100 mg l−1 kanamycin prevented the regeneration of non-transformed cells without being too toxic to the target explants; therefore this concentration was chosen as the selective agent.

Proliferation of cork oak somatic embryos in the presence of cefotaxime (C) and 100 mg l−1 kanamycin (K). Proliferation was measured as the number of embryos that maintained a white-yellowish colour, appeared to proliferate and did not become brown relative to the total number of embryos and is expressed as a percentage
About 2 months after transformation, white proliferating masses (Fig. 2b) were detected, and these were subsequently transferred to fresh selection medium. After a further 4 months of culture in selective medium, only the proliferating embryo masses were isolated—they were considered to be independent, putatively transgenic clones—and these were cultured in medium supplemented with kanamycin until sufficient material was available for molecular and histochemical analysis.
The susceptibility of cork oak somatic embryos to infection with three different A. tumefaciens strains (EHA105, LBA4404 and AGL1), all harbouring the pBINUbiGUSint plasmid, was also studied (Fig. 3). After 120 days of culture, the percentage of embryos showing recurrent proliferation varied significantly, depending on the strain used. Infection with the EHA105 and LBA4404 strains resulted in lower percentages of proliferation than infection with AGL1, with EHA105 stressing the embryos the most of the three strains tested. Thus it is not surprising that this latter strain of Agrobacterium did not render putative transformed embryos. The hypervirulent strain AGL1 was superior to LBA4404 and EHA105 in terms of putative transgenic secondary embryogenic proliferation. After 120 days of culture, 4±0.04% of the embryos initially inoculated with AGL1 pBINUbiGUSint proliferated in the presence of kanamycin, while only 0.6±0.05% of those initially inoculated with LBA4404 proliferated under the same condition. AGL1 is a disarmed derivative of C58, a hypervirulent bacterium that has been used successfully to infect various plant species (Hellens and Mullineaux 2000).
Following A. tumefaciens-mediated transformation, 34 independent putative transgenic embryo masses were assayed by PCR for the presence of the nptII and uidA genes. After a minimum of 120 days of culture in the presence of kanamycin, PCR analysis of the proliferating embryos showed that all clones were positive for nptII gene and approximately 98% of them for the uidA gene (Fig. 4). The negative uidA clones were discarded as probably having incomplete integration of the 3′ border of T-DNA.

Detection by PCR of the nptII (a) and GUS gene (uidA) (b). Lanes: 1, 11 DNA markers [PCR 100-bp low-molecular-weight ladder (Sigma, St. Louis, Mo.) (a), lambda DNA/EcoRI+HindIII (Promega, Madison, Wis. (b)], 2–7 transgenic clones obtained following inoculation with A. tumefaciens AGL1 pBINUbiGUSint, 8 transgenic clone obtained following inoculation with A. tumefaciens LBA4404 pBINUbiGUSint, 9 wild-type embryo clone, 10 pBINUbiGUSint plasmid containing the nptII and uidA genes
To avoid possible contamination and since amplification products could be due to residual bacteria, Hamill et al. (1990) suggested amplifying an area on the Ti-plasmid outside the T-DNA in order to indicate the presence of contaminating Agrobacterium. We therefore carried out a PCR analysis to detect a 202-bp band that corresponds to the virG gene of Agrobacterium. These clones were negative for virG (data not shown), suggesting that the cork oak transgenic clones were Agrobacterium-free.
The presence of GUS activity in embryos harbouring the uidA gene was investigated by histochemical assay (data not shown). The intensity of GUS staining was variable among the embryos. The distinctive GUS blue colouring appeared along the entire embryo surface, mainly in the axis. Control embryos did not develop in the presence of kanamycin nor did they not show any detectable GUS expression. In contrast, transgenic embryos proliferated in selective medium and expressed the uidA gene carrying the PIV2 intron (Vancanneyt et al. 1990), thereby demonstrating its functionality.
Further investigations on gene integration were then performed by Southern blot (Fig. 5). Genomic DNAs from putative transgenic lines were digested by HindIII (Fig. 5a), which liberates the uidA gene cassette, and by BglII, which has a unique restriction site located in the promoter of the uidA gene (Figs. 1, 5b). Southern blot analyses confirmed the presence of the uidA gene in transgenic cork oak genomes (Fig. 5a), since the hybridisation signals with the labelled probe were only associated with blotted DNA extracts known to be GUS-positive. This suggested the presence of the whole insert in all of the plants analysed because the uidA gene is located next to the left border of the T-DNA. No bands were observed in the control. Digestion with BglII allowed to estimate the number of gene copies using a HindIII-BglII fragment as probe (Fig. 5b). Clones in lanes 3, 4, 7 and 8 of Fig. 5b emerged independently from different transformation events, while clones shown in lanes 5 and 6 emerged independently from the same explant and were established separately. Nevertheless, these lines gave exactly the same hybridisation patterns, suggesting that were derived from the same transformation event. The number of inserts in the plants analysed ranged from 1 to 3. No bands were observed in the untransformed control plant.

Southern blot analysis of transformed embryo clones of cork oak. DNAs were digested with HindIII (a) or BglII (b) and probed using a [32P]-labelled HindIII (a) or HindIII-BglII (b) fragment containing the uidA gene prepared from pBINUbiGUSint plasmid. a Lanes: 1–4, 6, 8–11 DNA from independently transformed clones of cork oak, 5 untransformed cork oak plant, 7 potato plant harbouring the uidA gene (positive control). b Lanes 3–8 Transgenic clones of cork oak, 1 potato plant harbouring the uidA gene (positive control), 2 untransformed cork oak
As a final step kanamycin was removed from the medium to favour the maturation of embryos. Cefotaxime was removed as well and no Agrobacterium growth was observed, which confirmed the virG PCR results. Several independent transgenic embryos were successfully matured, germinated and transferred to soil following the protocol of Hernández et al. (2003a) (Fig. 2c, d). The presence of the transgenes in the acclimated plants was confirmed by PCR analysis (data not shown).
Although cork oak embryos can be produced following a unicellular pathway of regeneration preventing the formation of chimeric transgenic plants (Puigderrajols et al. 1996, 2000, 2001), the multicellular origin is the main way of secondary embryogenesis. Thus the formation of chimeric transgenic embryos is a problem that can be overcome using very active recurrent systems, such as the one described herein for cork oak. These systems of secondary embryogenesis coupled with culture on selective medium are useful for removing non-transgenic secondary embryos proliferating from chimeric primary somatic embryos (Merkle et al. 1990). Anatomical and histological studies have shown that cork oak primary somatic embryos show the initials of secondary embryogenesis within 2 weeks and cotyledonary secondary embryos can be seen within 20–30 days (Puigderrajols et al. 1996). After 3–4 months of subculture in the presence of kanamycin, we observed that all of the original putative transgenic embryogenic masses produced secondary embryos which were nptII-, uidA- and GUS-positive. These results support the hypothesis that chimeric embryos were not present in the last stages of culture. On the other hand, although the cork oak recurrent system is genetically quite stable and hence the risk of somaclonal variation is low (Gallego et al. 1997; Hornero et al. 2001), the specific culture conditions used to transform cells and to select transgenic embryos may induce changes that should be monitored.
The methodology described herein illustrates the possibility of transforming Quercus suber L. embryos obtained from selected mature plants using A. tumefaciens. This study is the first report of a protocol for obtaining transgenic plants from a mature selected cork oak tree. Studies to test this protocol with other selected cork oak genotypes are currently in progress. The results reported in this work demonstrate that transformation of selected mature trees is feasible, despite the scarcity of reports published to date on this topic.
Abbreviations
- BA:
-
N 6-Benzyladenine
- GUS:
-
β-Glucuronidase
- MSSH:
-
Expression-proliferation medium
- NAA:
-
α-Naphthaleneacetic acid
- nptII :
-
Neomycin phosphotransferase gene
- uidA :
-
β-Glucuronidase gene
References
An G, Ebert PR, Mitra A, Ha SB (1988) Binary vectors. In: Gelvin SB, Schilperoort RA, Verma, D-PS (eds) Plant molecular biology manual. Kluwer, Dordrecht, pp 1–19
Bevan M (1984) Binary Agrobacterium vectors for plan transformation. Nucleic Acids Res 12:8711–8721
Bueno MA, Astorga R, Manzanera JA (1992) Plant regeneration through somatic embryogenesis in Quercus suber. Physiol Plant 85:30–34
Christensen AH, Quail PH (1992) Maize polyubiquitin genes: genes, structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol Biol 18:675–689
Ecker P, Roshal S, Schell J, Willmitzer L (1986) Isolation and characterization of a light-inducible, organ-specific gene from potato and the analysis of its expression after tagging and transfer in tobacco and potato shoots. Mol Gen Genet 199:216–244
Fernández-Guijarro B, Celestino C, Toribio M (1994) Somatic embryogenesis in Quercus suber L. In: Pardos JA, Ahuja MR, Elena-Rossello R (eds) Biotechnology of trees. Investigación Agraria, Sistemas y Recursos Forestales, no 4, pp 105–110
Fernández-Guijarro B, Celestino C, Toribio M (1995) Influence of external factors on secondary embryogenesis and germination in somatic embryos from leaves of Quercus suber L. Plant Cell Tissue Organ Cult 41:99–106
Gallego FJ, Martínez I, Celestino C, Toribio M (1997) Testing somaclonal variations using RAPDs in Quercus suber L somatic embryos. Int J Plant Sci 158:563–567
Hamill J, Rounsley S, Spencer A, Todd G, Rhodes M (1990) The use of the polymerase chain reaction to detect specific sequences in transformed plant tissues. In: Nijkamp HJS, Van der Plas LHW, Van Aartijk (eds) Progress in plant cellular molecular biology. Kluwer, Dordrecht pp 183–188
Hellens R, Mullineaux P (2000) A guide to Agrobacterium binary Ti vectors. Trends Plant Sci 5:446–451
Hernández I, Celestino C, Martínez I, Manjón JL, Díez J, Fernández-Guijarro B, Toribio M (2001) Cloning mature cork oak (Quercus suber L.) trees by somatic embryogenesis. Melhoramento 37:50–57
Hernández I, Celestino C, Alegre J, Toribio M (2003a) Vegetative propagation of Quercus suber L. by somatic embryogenesis. II. Plant regeneration from selected cork oak trees. Plant Cell Rep 21:765–770
Hernández I, Celestino C, Toribio M (2003b) Vegetative propagation of Quercus suber L. by somatic embryogenesis. I. Factors affecting the induction in leaves from mature cork oak trees. Plant Cell Rep 21:759–764
Hoekema A, Hirsch PR, Hooykaas PJJ, Schilperoort RA (1983) A binary vector strategy based on separation of vir and T-region of the Agrobacterium tumefaciens Ti plasmid. Nature 303:179–180
Hood EE, Clapham DH, Ekberg I, Johanson T (1990) T-DNA presence and opine production in tumors of Picea abies (L.) Karst induced by Agrobacterium tumefaciens A281. Plant Mol Biol 14:111–117
Hornero J, Martinez I, Celestino C, Gallego FJ, Torres V, Toribio M (2001) Early checking of genetic stability of cork oak somatic embryos by AFLP analysis. Int J Plant Sci 162:827– 833
Humara J, Ordás RJ (1999) The toxicity of antibiotics and herbicides on in vitro adventitious shoot formation on Pinus pinea L. cotyledons. In Vitro Cell Dev Biol Plant 35:339–343
Humara J, Martín MS, Parra F, Ordás RJ (1999) Improved efficiency of uidA gene transfer in stone pine (Pinus pinea) cotyledons using a modified binary vector. Can J For Res 29:1627–1632
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene system. Plant Mol Biol Rep 5:387–405
Lazo GR, Stein PA, Ludwig RA (1991) A DNA transformation-competent Arabidopsis genomic library in Agrobacterium. Biotechnology 9:963–967
Manzanera JA, Astorga R, Bueno MA (1993) Somatic embryo induction and germination in Quercus suber L. Silvae Genet 42:90–93
Merkle SA, Dean J FD (2000) Forest tree biotechnology. Curr Opin Biotechnol 11:298–302
Merkle SA, Parrot WA, Williams EG (1990) Applications of somatic embryogenesis and embryo cloning. In: Bhojwani SS (ed) Plant tissue culture: applications and limitations. Elsevier, Amsterdam, pp 67–101
Nauerby B, Billing K, Wyndaele R (1997) Influence of the antibiotic timentin on plant regeneration compared to carbenicillin and cefotaxime in concentrations suitable for elimination of Agrobacterium tumefaciens. Plant Sci 123:169–177
Peña L, Séguin A (2001) Recent advances in the genetic transformation of trees. Trends Biotechnol 19:500–506
Puigderrajols P, Fernández-Guijarro B, Toribio M, Molinas M (1996) Origin and early development of secondary embryos in Quercus suber L. Int J Plant Sci 157:674–684
Puigderrajols P, Celestino C, Suils M, Toribio M, Molinas M (2000) Histology of organogenic and embryogenic responses in cotyledons of somatic embryos of Quercus suber L. Int J Plant Sci 161:353–362
Puigderrajols P, Mir G, Molinas (2001) Ultrastructure of early secondary embryogenesis by multicellular and unicellular pathways in cork oak (Quercus suber L.). Ann Bot 87:179–189
Roest S, Brueren HGMJ, Evers PW, Vermeer E (1991) Agrobacterium-mediated transformation of oak (Quercus robur L.). Acta Hortic 289:259–260
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Seabra RC, Pais MS (1997) Genetic transformation of European chestnut. Plant Cell Rep 17:177–182
Seabra RC, Pais MS (1999) Genetic transformation of European chestnut (Castanea sativa Mill.) with genes of interest. Acta Hortic 494:407–414
Vancanneyt G, Schmidt R, O’Connor-Sánchez A, Willmitzer L, Rocha-Rosa M (1990) Construction of an intron-containing marker gene: splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium-mediated plant transformation. Mol Gen Genet 220:245–250
Wilhelm E (2000) Somatic embryogenesis in oak (Quercus spp.). In Vitro Cell Dev Biol Plant 36:349–357
Wilhelm E, Burg A, Berenyi M, Endemann M, Rodler R (1996) Plantlet regeneration via somatic embryogenesis and investigations on Agrobacterium tumefaciens mediated transformation of oak (Quercus robur). In: Ahuja MR, Boerjan W, Neale DB (eds) Somatic cell genetics and molecular genetics of trees. Kluwer, Dordrecht, pp 119–124
Zobel BJ, Talbert JT (1984) Vegetative propagation. In: Applied forest tree improvement. Wiley, New York, pp 309–344
Acknowledgements
The authors thank Dr. J.M. Martín-Alonso for his technical support and Dr. J. Fernández-Humara for revision of the manuscript. R. Álvarez is supported by a FICYT research fellowship funded by Plan Investigación, Desarrollo Tecnológico e Innovación de Asturias 2001–2004 of the Gobierno del Principado de Asturias.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Peña
Rights and permissions
About this article
Cite this article
Álvarez, R., Alonso, P., Cortizo, M. et al. Genetic transformation of selected mature cork oak (Quercus suber L.) trees. Plant Cell Rep 23, 218–223 (2004). https://doi.org/10.1007/s00299-004-0810-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0810-2