
Abstract
Transgenic plants are potentially safe and inexpensive vehicles to produce and mucosally deliver protective antigens. However, the application of this technology is limited by the poor response of the immune system to non-particulate, subunit vaccines. Co-delivery of therapeutic proteins with carrier proteins could increase the effectiveness of the antigen. This paper reports the ability of transgenic Arabidopsis thaliana plants to produce a fusion protein consisting of the B subunit of the Escherichia coli heat-labile enterotoxin and a 6 kDa tuberculosis antigen, the early secretory antigenic target ESAT-6. Both components of the fusion protein were detected using GM1-ganglioside-dependent enzyme-linked immunosorbant assay. This suggested the fusion protein retained both its native antigenicity and the ability to form pentamers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The technology of edible vaccines has vast potential to help the ailing or non-existent health systems of developing countries and to revolutionize vaccine delivery within veterinary medicine and the developed world health systems. However, a number of limitations must be resolved before this technology can be applied. One such limitation is the unresponsiveness of the mucosal immune system to small, non-particulate antigens (such as many candidate subunit vaccines). Co-administration with an adjuvant or targeting protein can increase the mucosal immunogenicity of proteins or peptides otherwise imperceptible to the mucosal immune system.
One such carrier protein for subunit vaccines is the heat-labile toxin (LT) of enterotoxigenic Escherichia coli (ETEC). This toxin consists of an enzymatically active A subunit (LTA), and a non-toxic B subunit (LTB). The B subunit forms a donut-shaped pentamer that targets the holotoxin to the mucosal lymphoid tissues by binding to the GM1 gangliosides found on the surface of mucosal epithelial cells (Hol et al. 1995). Hence, an antigen can be delivered across the epithelial cells lining the mucosal tracts to the underlying mucosal-associated lymphoid tissue by chemically linking or genetically fusing the antigen to LTB in a manner that allows pentamer formation (Liljeqvist et al. 1997). LTB has been extensively studied as a carrier protein within bacterial expression systems (Bagdasarian et al. 1999; Lipscombe et al. 1991; O’Dowd et al. 1999). However, only one plant-derived LTB fusion has been reported (Walmsley et al. 2003), which investigated an addition of only six amino acids to the carboxy terminus of LTB.
Tuberculosis (TB) is one of the most widespread diseases of mankind and animals. While Mycobacterium tuberculosis is the main pathogen of man, M. bovis afflicts one of the broadest host ranges of all known pathogens, including hosts such as cattle, bison, buffalo, antelope, badgers, deer, ferrets and brushtail possum. M. bovis is also responsible for approximately 6% of total human TB deaths through zoonosis. Tuberculosis is currently controlled industrially by either whole herd slaughter upon detection of an infected animal or whole herd diagnostic testing and slaughter of reactor animals.
Recent reports (Horwitz et al. 1995, 2000) have demonstrated that immunization with purified M. tuberculosis major extracellular proteins induces substantial protective immunity in guinea pig against aerosol challenge with the highly virulent Erdman strain of M. tuberculosis. A promising candidate amongst the major extracellular proteins is the low molecular-weight protein termed ESAT-6 (early secretory antigenic target—6 kDa). The ESAT-6 protein is expressed only by virulent M. bovis and M. tuberculosis strains and is a dominant target for cell-mediated immunity in the early phase of tuberculosis in various animal models (Brandt et al. 1996; Pollock and Andersen 1997). In mice, vaccination with ESAT-6 delivered in a combination of monophosphoryl lipid A and dimethyl dioctadecylammonium bromide elicited a strong ESAT-6-specific T-cell response and protective immunity (Brandt et al. 1996). As the first step in the development of a plant-derived, mucosally delivered and targeted, subunit tuberculosis vaccine for animals, the production of an LTB/ESAT-6 fusion protein in a model plant species, Arabidopsis thaliana is reported.
Materials and methods
ESAT-6 design and assembly
A plant-optimized ESAT-6 coding region was designed from the native sequence (GenBank accession number U34848) to maintain the amino acid sequence of the native gene yet contain codons preferred by plant genes (Wada et al. 1990). Convenient BbsI and KpnI restriction sites 5′ and 3′, respectively, of the ESAT-6 coding region were included for ease of subsequent cloning. The amino acid sequence of the LTB/ESAT-6 fusion protein was analyzed using PSORT (http://psort.nibb.ac.jp) and EXPASy Proteomics tools (http://expasy.ch/tools/) for the prediction of protein localization in the plant cell.
Both strands of the plant-optimized coding sequence were divided into overlapping oligonucleotides about 50 bp in length before assembly by overlap extension PCR, as described by Stemmer et al. (1995). The synthetic gene was cloned into a commercial PCR cloning vector, pCR-XL-TOPO (Invitrogen, Grand Island, N.Y.), to create pTOPOESAT-6 and sequenced by the dideoxy chain termination method (Sanger et al. 1977).
Genetic fusion of ESAT-6 and LTB and insertion into a plant expression cassette
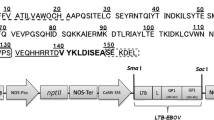
The plasmid pLTB-L (Walmsley et al. 2003) was prepared for insertion of ESAT-6 by serial digestion with BbsI and KpnI and dephosphorylation with calf intestinal phosphatase. The BbsI-KpnI fragment containing the ESAT-6 gene was excised from pTOPOESAT-6 and cloned downstream of the LTB gene within pLTB-L. The LTB/ESAT-6 fusion gene was digested with NcoI and KpnI and cloned into the plant expression cassette pIBT210 (Haq et al. 1995), creating pAW8. Spontaneous mutation during cloning resulted in a clone containing a single amino acid linker (asparagine). This clone was named pAWSmL8. The fused coding regions within pAW8 and pAWSmL8 were cloned downstream of the cauliflower mosaic virus 35S promoter with a double enhancer region (CaMV 35S) and upstream of the soybean vegetative storage protein (VSP) 3′ terminator region (Fig. 1). Clones were verified through PCR and sequence analysis then inserted into the HindIII and EcoRI sites of the binary vector pGPTV.kan (Becker et al. 1992), creating pAWBin8 and pAWBinSmL8. Both binary vectors contain the nptII gene for selection of transformed plant cells.

Diagrammatic representation of pAW8. Rb, Lb Right and left T-DNA border repeats, respectively; CaMV 35S 5′ untranslated region of cauliflower mosaic virus 35S promoter with double enhancer, TEV 5′ untranslated region of tobacco etch virus, LTB synthetic B subunit of Escherichia coli heat-labile enterotoxin gene, L peptide linker, ESAT-6 synthetic early secretory antigenic target—6 kDa—gene, VSP 3′ untranslated region of the soybean vegetative storage protein
Arabidopsis thaliana transformation
Within this paper, subscript text used to describe a transgenic plant line, for example T1, indicates the number of sexual cycles that have occurred after the transformation event. A. thaliana was chosen for this study due to the relative ease, speed and reliability of the transformation protocol. The plasmids pAWBin8, pAWBinSmL8 and TH110 (containing the LTB gene; Haq et al. 1995) were prepared from cultures of E. coli DH5α and electroporated individually into Agrobacterium tumefaciens LBA4404 (Hoekema et al. 1983). Floral dip Agrobacterium-mediated transformation of A. thaliana was performed as described by Clough and Bent (1998). The resulting seeds were collected and selected for transgenic lines by germination and growth on medium containing 50 µg/ml kanamycin. Seeds from the pAWBin8 and pAWBinSML8 T1 transformants expressing the highest level of LTB were germinated. The segregation ratio in the T2 progeny was roughly 3:1 kanamycin-resistant:kanamycin-susceptible seedlings.
Nucleic acid extraction from T1 plants
Fresh leaves from T1 plants germinated and grown in GA-7 boxes (Magenta, Chicago, Ill.) were used for genomic DNA extraction. About 100 mg leaf material was collected and snap frozen with liquid nitrogen before being crushed to a fine powder using a hardware nail. Genomic DNA was then prepared using a CTAB extraction protocol (Hwang and Kim 2000). Total RNA was extracted using the RNAqueous kit according to the manufacturer’s instructions (Ambion, Huntingdon, Cambridgeshire, UK).
Amplification and detection of the transgene by PCR
The genomic DNA of putative transgenic plants was used as template in a transgene-specific PCR using primers TEV (tobacco etch virus; 5′-GCATTCTACTTCTATTGCAGC) and VSP (5′-GTGCATATCAGCATAC). Using these primers, an amplicon of 813 bp or 795 bp was expected. The presence or absence of the seven amino acid linker was also verified in pAWBin8 and pAWBinSmL8 transformations by a linker-specific PCR using primers Link (5′ GATCCACATGTTCCTAAC) and VSP. The same genomic DNA samples were used in both transgene-specific and linker-specific PCR. A band of 352 bp should be present only if the seven amino acid linker is present in the transforming construct. Samples were used in a standard PCR using an annealing temperature of 55°C. A negative control of wild type A. thaliana genomic DNA and positive control of pAW8 or pAWSmL8 plasmid were included in each experiment. PCR samples were run on a 1% (TAE) agarose gel against a 1 kb ladder (Gibco BRL, Rockville, Md.).
Northern analysis
Total RNA samples from pAWBin8 transformants and wild type plants were separated on a gel and transferred to a membrane as described in Sambrook et al (1989). A PCR-labeled probe was prepared using primers TEV and VSP on a pAW8 template. Digoxygenin (DIG)-labeled dCTP was incorporated into the 813 bp amplicon according to the manufacturer’s instructions (PCR DIG probe synthesis kit, Roche, Mannheim, Germany).
Hybridization bottles and 10 ml DIG Easy Hyb (Roche) per membrane were pre-warmed to 45°C in a hybridization oven. Membranes were prehybridized for at least 90 min and hybridized overnight with 5 μl probe added per milliliter DIG Easy Hyb. Post-hybridization washes and detection were performed according to the manufacturer’s instructions (DIG wash and block buffer set and DIG luminescent detection kit; Roche). Labeled membranes were visualized after exposure to film.
Protein extraction and antigen analysis
Western blot analysis
Crude protein extracts were prepared by homogenizing fresh leaves in phosphate-buffered saline (PBS) (1 ml PBS/g leaves) in a QBiogene Fast Prep machine (Carlsbad, Calif.). Insoluble material was removed by centrifugation at 14,000 rpm in an Eppendorf 5415C microcentrifuge at 4°C for 5 min. For ESAT-6 analysis, the resulting sample supernatants were kept on ice during analysis and subsequently stored at −80°C. For LTB analysis, 10 μl aliquots were removed from a bulk crude protein extract at 10 min intervals starting at 0 and ending at 60 min. All of the samples were boiled for 10 min and placed on ice. Western blot detection of LTB and its derivatives was performed as per Walmsley et al. (2003). The anti-LTB antiserum contained polyclonal antibodies raised in rabbits after injection with recombinant, bacterial LTB. Western blot detection of ESAT-6 and its derivatives was performed in a manner similar to that of LTB except that the primary antibody was anti-ESAT-6 antiserum (TB Research Materials and Vaccine Testing contract NIH, NIAID No1 AI-75320, Colorado State University) diluted 1:5,000 in PBSTM (1% dry milk + PBS +0.1% Tween-20). Purified, recombinant, bacterial ESAT-6 was supplied by the TB Research Materials and Vaccine Testing Contract at Colorado State University and used as the positive control. The anti-ESAT-6 antiserum contained polyclonal antibodies raised in rabbits after injection with recombinant bacterial ESAT-6.
Enzyme-linked immunosorbant assay analysis
Frozen A. thaliana plant samples were ground using a QBiogene Fast Prep machine and resuspended in extraction buffer (PBS +5% dry milk, 0.1% Triton X-100, 10 μg/ml leupeptin; 1 ml buffer/g sample). Insoluble materials were pelleted by centrifugation at 14,000 rpm in an Eppendorf 5415C microcentrifuge at 4°C for 5 min. The supernatant was kept on ice during analysis then stored at −80°C. Ganglioside-dependent enzyme-linked immunosorbant assay (ELISA) (Haq et al. 1995) was performed on two samples of varying dilutions in PBSTM of each extract using a primary antibody of either 1:1,000 anti-LTB antiserum or anti-ESAT-6 antiserum in PBSTM.
Although ganglioside-dependent ELISA was used for the detection of ESAT-6 within the plant samples, the ESAT-6 standards were directly bound to the plates. The standard for ESAT-6 detection was a recombinant bacterial form provided by the TB Research Materials and Vaccine Testing contract NIH, NIAID No1 AI-75320, Colorado State University.
Fusion protein deglycosylation and cleavage
In an attempt to identify bands revealed in western analysis, crude extracts from freeze-dried leaves were partially purified using galactose affinity chromatography (Walmsley et al. 2003), digested with 1 or 2 mU N-glycosidase A from almond (Calbiochem, La Jolla, Calif.) for 24 h at 37°C, and examined by anti-LTB western analysis as previously described. Protein extracts in extraction buffers A (PBS; 1 ml/g leaf material); B [PBS, complete protease inhibitor EDTA-free (Roche), 1 mM EDTA; 1 ml/g plant material]; C (50 mM NaH2PO4 pH 6.6, 100 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 10 μg/ml leupeptin; 1 ml/g leaf material); and D (50 mM NaH2PO4 pH 6.6, 100 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, complete protease inhibitor EDTA-free; 1 ml/g leaf material) were compared through anti-LTB western analysis.
Effect of freeze-drying on antigen conformation and stability
Leaf material from T2 Arabidopsis plants was freeze-dried for a minimum of 72 h with a maximum shelf temperature of 20°C. The dried leaf material was powdered, pooled, and stored in a dry environment at −20°C. Pooled, freeze-dried leaf material was analyzed in an anti-LTB western as previously described for fresh materials as well as anti-LTB ELISAs at 0, 2 and 4 months.
Results and discussion
In designing the plant-optimized ESAT-6 gene, 55% of the codons were replaced and the G+C content was decreased from 60% to 47% in order to match codon usage by dicots, eliminate potential polyadenylation signals and eliminate sequences that might cause mRNA destabilization and splicing. As previously stated, plant-derived LTB is directed to the endoplasmic reticulum (ER) through its cleavable amino-terminal signal peptide, which targets LTB to the periplasmic space in bacteria (Mason et al. 1998). Using PSORT (prediction of protein sorting signals and localization sites in amino acid sequences) to evaluate the fusion protein amino acid sequence, the fusion protein was predicted to have a cleavable amino acid signal peptide (Fig. 2). The fusion protein was not thought to be a transmembrane protein or targeted to the mitochondria, chloroplast, vacuole or peroxisomes. Instead the LTB/ESAT-6 protein was predicted to travel through the vesicular pathway passing from the ER to the Golgi bodies before being secreted outside the plant cell. The EXPASy Proteomics program also predicted the fusion protein to be secretory and to have a 21 amino acid signal peptide (Emanuelsson et al. 2000; Nielsen et al. 1997).

Signal peptide and N-glycosylation analysis of the LTB/ESAT-6 fusion protein. Amino acids in single letter format: lower case LTB gene, upper case ESAT-6, underlined linker, * translation stop site, italics signal peptide, bold possible N-linked glycosylation sites
Nucleic acid analysis
An 813-bp or 795-bp band, corresponding to the expected size of the fusion gene, was amplified by PCR from DNA samples from kanamycin-resistant plants transformed with the binary vector pAWBin8 or pAWBinSmL8, respectively (Fig. 3a, b). No band was amplified from DNA samples from non-transformed plants. Using the linker-specific PCR primers, only the pAW8 plasmid control displayed the expected band size of 352 bp (Fig. 3c). This demonstrated that the putative transgenic plants germinated on kanamycin did indeed possess the desired gene with (pAWBin8) or without (pAWBinSmL8) the linker.

PCR analysis of genomic DNA extracts from Arabidopsis thaliana plants. Lanes: L Gibco-BRL 1 kb ladder, 1–5 PCR using putative transgenic A. thaliana genomic DNA from different individuals as template, P vector DNA as template, N wild type A. thaliana genomic DNA as template. a Transgene-specific PCR of pAWBin8-transformed plants; P pAW8. b Transgene-specific PCR of pAWBinSmL8-transformed plants; P pAWSmL8. c Linker-specific PCR of pAWBinSmL8-transformed plants; P pAW8, Pl pAWSmL8
Northern analysis of total RNA demonstrated no band in the wild type negative control. However a band was visualized around the predicted size (1,000 bp) in different transgenic A. thaliana lines (Fig. 4). The foreign gene was therefore transcribed correctly with no detectable cleavage or degradation products. Because the gene was codon-optimized for plant expression, it is unlikely expression would be due to residual Agrobacterium in the tissue.

Northern analysis of wild type and pAWBin8 transgenic A. thaliana lines. Lanes: N Total RNA extracted from a wild type A. thaliana plant, 1–3 total RNA extracted from three transgenic A. thaliana lines
Analysis of fusion protein expression
Anti-ESAT-6 western blot analysis revealed the recombinant, bacterial ESAT-6 positive control to run at about 11 kDa (Fig. 5). The band detected at about 11 kDa was thought to be a combination of ESAT-6 monomers and dimers running at the gel front. The additional larger bands within the ESAT-6 positive control lane were thought to be multimers of ESAT-6 or non-specific binding to contaminants (Fig. 5a, b; lane P). In the native gel, the negative control (Fig. 5a; lane N) displayed non-specific bands around 55 and larger than 150 kDa, while the plant-derived ESAT-6 sample (Fig. 5a; lane T) had bands running at about 45, 70, and higher than 150 kDa. The three bands were thought to be multimers of the fusion protein. The gel using denaturing conditions displayed a non-specific band at about 22 kDa within the negative control and the three transgenic A. thaliana lines. However, a band running at about 30 kDa was seen only in the transgenic lines.

Anti-ESAT-6 western blot analysis of the fusion protein in transgenic tissues. Native samples were neither boiled nor run in the presence of DTT. Samples run under denaturing conditions were boiled for 10 min in the presence of DTT. a Non-boiled, native samples. Lanes: P Native, purified, bacterial ESAT-6; T native total protein from a transgenic line of A. thaliana; B blank lane; N native total protein from A. thaliana wild type plant. b Boiled, denatured samples. Lanes: N Total protein from A. thaliana wild type plants in the presence of DTT, P 500 ng purified bacterial ESAT-6, 1–3 total protein from A. thaliana transgenic plants. Band sizes are given in kilodaltons
Anti-LTB western blot analysis revealed a nonspecific band of about 18 kDa present in both the wild type A. thaliana line and the lines expressing the fusion protein and LTB (Fig. 6a). Two bands, about 24 and 12 kDa, were observed in the bacterial LTB control (Fig. 6, lane B). The 12 kDa band was identified as LTB monomers, the 24 kDa band is thought to be the result of non-specific binding to contaminants. A band of 12 kDa was present in the A. thaliana line expressing LTB alone (Fig. 6a, lane P). A band corresponding to about 30 kDa and a doublet centered at 14 kDa were observed within the transgenic A. thaliana line. Since a band at 30 kDa was also observed in anti-ESAT-6 western blot analysis (Fig. 5b) this band was designated the LTB/ESAT-6 fusion protein. After 20 min at room temperature, a band was observed at 25 kDa, and after 30 min at 16 kDa. Anti-LTB western analysis of other T2 plants demonstrated the same banding pattern (data not shown).

Anti-LTB western blot analysis. a Protein extraction from one transformed individual using phosphate-buffered saline (PBS) as the extraction buffer. The protein extract was left at room temperature for 0, 10, 20, 30, 40, 50 or 60 min. Lanes: B Purified bacterial LTB (50 ng), P total protein from A. thaliana transgenic plant expressing LTB alone, N total protein from A. thaliana wild type plants. b Effect of protease inhibitors on banding pattern in western analysis of freeze-dried, partially purified, A. thaliana extracts. Lanes: B Purified bacterial LTB (20 ng); A1, A2 transgenic extracts in buffer A; B1, B2 transgenic extracts in buffer B; C transgenic extracts in buffer C; D transgenic extracts in buffer D (see Materials and methods for buffer compositions). Band sizes are given in kilodaltons
To determine if additional bands observed in anti-LTB western analysis were degraded, or represented cleavage or glycosylated transgene products, we incubated plant extracts with N-glycosidase A (from almond) or protease inhibitors in two experiments. In the deglycosylation, anti-LTB western, lanes containing deglycosylated, partially purified, LTB fusion protein and possible cleavage products did not differ from lanes containing corresponding non-treated controls (data not shown). The same was observed when comparing extracts with and without added protease inhibitors (Fig. 6b). The 14 kDa-centered doublet plus the 16 and 25 kDa bands in Fig. 5 were therefore thought to be cleavage products of the fusion protein (30 kDa). Since the bands were present in samples that had protease inhibitors present, the cleavage was thought to have occurred as the fusion protein circulated through the ER system. Given that the 14 kDa doublet was also seen in anti-LTB western analysis of the fusion protein with only a single amino acid linker (Fig. 7a) it was determined that the seven amino acid linker was not the cause of cleavage of the fusion protein. This experiment was also able to discern the difference in protein size due to the loss of six amino acids in the pAWBinSmL8 transformants (Fig. 7a, b).

Western blot analysis of A. thaliana plant lines transformed with pAWBin8 and pAWBinSmL8. a Anti-LTB immunodetection. b Anti-ESAT-6 immunodetection. Lanes: L Protein extracts from plants transformed with the pAWBin8 (seven amino acid linker), SmL protein extracts from plants transformed with the pAWBinSmL8 (one amino acid linker). Band sizes are given in kilodaltons
ELISA analysis
Previous studies of bacterial expression of LTB fusions were inconclusive as to whether the process reduces the ability to form pentamers (Clements 1990; Jagusztyn-Krynicka et al. 1993; Sandkvist et al. 1987). Fusions to LTB should therefore be studied on a case-by-case basis. Analysis of variance determined the signals from transformed plant lines in anti-ESAT-6 and anti-LTB ELISAs as significantly higher than the readings gained for the protein extract from wild type A. thaliana (α =0.05, Fig. 8). The carboxyl terminus extension from LTB of a seven amino acid linker and ESAT-6 therefore did not hamper pentamer formation or the ability of the fusion protein to bind to GM1 gangliosides. There was no significant difference between expression levels (anti-LTB or -ESAT-6 ELISAs) between pAWBin8 or pAWBinSmL8 transformants (α =0.05). Since polyclonal antisera raised against recombinant bacterial forms of the LTB and ESAT-6 proteins were able to detect the plant-derived fusion protein under denaturing and native conditions it suggested the fusion retained its native antigenicity.

GM1 enzyme-linked immunosorbant assay (ELISA) detection of a LTB protein levels and b ESAT-6 levels in transgenic A. thaliana plants. WT Total protein extract from wild type A. thaliana plants; ESAT-6 250 ng purified, recombinant, bacterial ESAT-6; LTB protein extract from A. thaliana line expressing LTB alone; SmL A. thaliana line transformed with pAWBinSmL8
Within the anti-ESAT-6 ELISA, two additional negative controls were tested along side wild type plant crude protein extract. Purified, recombinant ESAT-6 was added to GM1 ganglioside-bound wells to demonstrate the inability of ESAT-6 to bind to gangliosides without LTB, whilst protein extract from an A. thaliana line expressing LTB alone was added to ganglioside-bound wells to test whether the anti-ESAT-6 antibodies had any affinity to LTB. Since analysis of variance did not determine significant difference between these controls and the OD of wild type A. thaliana plant protein extracts (α =0.05, Fig. 8b) we are confident that the observed ODs for the transgenic lines are due to ganglioside-bound LTB/ESAT-6 fusion protein.
The ESAT-6 standards were bound directly to the plate and not to GM1 gangliosides (as with the fusion protein). The ESAT-6 standards were therefore deemed an inaccurate measure of fusion protein concentration in the plant extracts. Instead, the concentration of fusion protein in the plant materials was based on LTB content. This varied from 22 to 49 µg/g fresh weight. As previously discussed, however, about half of the LTB signal was due to the 14, 16 and 25 kDa LTB/ESAT-6 cleavage products. Hence the fusion protein concentrations were estimated to be half of the demonstrated LTB content, varying between 11 and 24.5 µg/g fresh weight.
Effect of freeze-drying on antigen content
Anti-LTB western analysis of freeze-dried Arabidopsis materials displayed no difference in banding pattern compared to non-processed materials (Fig. 6b). However, the antigen content was concentrated roughly 7-fold (Figs. 8a, 9). Anti-LTB ELISA analysis over time also displayed no loss in LTB content or activity (Fig. 9) when materials were stored in a dry environment at −20°C. The pooling of processed Arabidopsis samples thus provided a batch of plant material with concentrated, antigenically active fusion protein of uniform concentration that displayed a −20°C shelf life of at least 4 months.

Stability of LTB content in freeze-dried, transgenic A. thaliana materials using GM1-dependent ELISA. Error bars Standard error of the mean
Conclusion
The ability of transgenic A. thaliana plants to express a fusion protein consisting of LTB and ESAT-6 was investigated. Immunoblot analysis of crude protein extracts from A. thaliana plants transformed with the fused coding regions revealed that the plant-derived LTB/ESAT-6 monomer has an apparent molecular weight of 30 kDa, while both the bacterial and the Arabidopsis-synthesized LTB monomer have a molecular weight of 12 kDa. The ability to detect the LTB fusion protein through anti-ESAT-6 and anti-LTB GM1 ganglioside-dependent ELISA, in addition to native anti-ESAT-6 western analysis, indicated that the extension of ESAT-6 plus linker to the LTB subunit carboxyl terminus did not prevent LTB pentamer formation and that both components of the fusion protein retained antigenicity. A one or seven amino acid linker was inserted between the LTB and ESAT-6 proteins to decrease the chance of the ESAT-6 extension interfering with LTB pentamer assembly. Since no significant difference was found between the expression levels of both forms of the fusion protein (one and seven amino acid linker) using GM1 ganglioside dependent anti-LTB or anti-ESAT-6 ELISA, it was concluded that a linker is required neither for correct processing and assembly of the fusion protein nor for maintenance of its antigenicity.
To standardize and concentrate the antigen in plant material, we pooled and freeze-dried the Arabidopsis material. Freeze-drying is an established food processing technology that is relatively simple and inexpensive. As it increases both antigen concentration and antigen shelf-life it should prove useful in developing plant-derived antigens as traditional vaccine alternatives.
Abbreviations
- ELISA :
-
Enzyme linked immunosorbant assay
- ESAT-6 :
-
Early secretory antigenic target (6 kDa)
- ETEC :
-
Enterotoxigenic Escherichia coli
- LTB :
-
B subunit of E. coli heat-labile enterotoxin
References
Bagdasarian MM, Nagai M, Frey J, Bagdasarian M (1999) Immunogenicity of Actinobacillus ApxIA toxin epitopes fused to the E. coli heat-labile enterotoxin B subunit. Vaccine 17:441–447
Becker D, Kemper E, Schell J, Masterson R (1992) New plant binary vectors with selectable markers located proximal to the left T-DNA border. Plant Mol Biol 20:1195–1197
Brandt L, Oettinger T, Holm A, Andersen AB, Andersen P (1996) Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J Immunol 157:3527–3533
Clements JD (1990) Construction of a nontoxic fusion peptide for immunization against Escherichia coli strains that produce heat-labile and heat-stable enterotoxins. Infect Immun 58:1159–1166
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1006
Haq TA, Mason HS, Clements JD, Arntzen CJ (1995) Oral immunization with a recombinant bacterial antigen produced in transgenic plant. Science 268:714–716
Hwang SK, Kim YM (2000) A simple and reliable method for preparation of cross-contamination-free plant genomic DNA for PCR-based detection of transgenes. J Biochem Mol Biol 33:537–546
Hoekema A, Hirsch PR, Hooykaas PJJ, Schilperoort RA (1983) A binary plant vector strategy based on separation of vir- and T-region of the Agrobacterium tumefaciens Ti Plasmid. Nature 303:179–180
Hol WGJ, Sixma TK, Merritt EA (1995) Structure and function of E. coli heat-labile enterotoxin and cholera toxin B pentamer. In: Moss J, Iglewski B, Vaughan M, Tu AT (eds) Bacterial toxins and virulence factors in disease. Dekker, New York, pp 185–223
Horwitz MA, Lee BE, Dillon BJ, Harth G (1995) Protective immunity against tuberculosis induced by vaccination with major extracellular proteins of Mycobacterium tuberculosis. Proc Natl Acad Sci USA 92:1530–1534
Horwitz MA, Dillon BJ, Galic SM (2000) Recombinant bacillus Calmette-Guerin (BCG) vaccines expressing the Mycobacterium tuberculosis 30-kDa major secretory protein induce greater protective immunity against tuberculosis than conventional BCG vaccines in a highly susceptible animal model. Proc Natl Acad Sci USA 97:13853–13858
Jagusztyn-Krynicka EK, Clark-Curtiss JE, Curtiss R 3rd (1993) Escherichia coli heat-labile toxin subunit B fusions with Streptococcus sobrinus antigens expressed by Salmonella typhimurium oral vaccine strains: importance of the linker for antigenicity and biological activities of the hybrid proteins. Infect Immun 61:1004–1015
Liljeqvist S, Stahl S, Andreoni C, Binz H, Uhlen M, Murby M (1997) Fusions to the cholera toxin B subunit: influence on pentamerization and GM1 binding. J Immunol Methods 210:125–135
Lipscombe M, Charles IG, Roberts M, Dougan G, Tite J, Fairweather NF (1991) Intranasal immunization using the B subunit of the Escherichia coli heat-labile toxin fused to an epitope of the Bordetella pertussis P.69 antigen. Mol Microbiol 5:1385–1392
Mason HS, Haq TA, Clements JD, Arntzen CJ (1998) Edible vaccine protects mice against Escherichia coli heat-labile enterotoxin (LT): potatoes expressing a synthetic LT-B gene. Vaccine 16:1336–1343
Nielsen H, Engelbrecht J, Brunak S, von Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6
O’Dowd AM, Botting CH, Precious B, Shawcross R, Randall RE (1999) Novel modifications to the C-terminus of LTB that facilitate site-directed chemical coupling of antigens and the development of LTB as a carrier for mucosal vaccines. Vaccine 17:1442–1453
Pollock JM, Andersen P (1997) Predominant recognition of the ESAT-6 protein in the first phase of interferon with Mycobacterium bovis in cattle. Infect Immun 65:2587–2592
Sandkvist M, Hirst TR, Bagdasarian M (1987) Alterations at the carboxyl terminus change assembly and secretion properties of the B subunit of Escherichia coli heat-labile enterotoxin. J Bacteriol 169:4570–4575
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., pp 7.43–7.48
Sanger F, Nicklen S, Coulson AR (1977) DNA Sequencing with chain terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164:49–53
Wada K, Aota S, Tsuchiya R, Ishibashi F, Gojobori T, Ikemura T (1990) Codon usage tabulated from the GenBank genetic sequence data. Nucleic Acids Res 18:2367–2411
Walmsley AM, Alvarez ML, Jin Y, Kirk DD, Lee SM, Pinkhasov J, Rigano MM, et al (2003) Expression of the B subunit of Escherichia coli heat-labile enterotoxin as a fusion protein in transgenic tomato. Plant Cell Rep 21:1020–1026
Acknowledgements
We thank Dr. Hugh Mason of The Boyce Thompson Institute for supply of the basic vectors pLTB-L, TH110 and pGPTV.kan, Benchmark Biolabs Inc. for supply of the LTB standards and primary antibodies and the TB Research Materials and Vaccine Testing Contract at Colorado State University for supply of the ESAT-6 standards and primary antibodies. We thank Dr. Lokesh Joshi for his time and expertise in protein glycosylation. This work was supported by the internal competitive grant scheme of the Boyce Thompson Institute for Plant Research and Dow AgroSciences LLC. Dr. M. Lucrecia Alvarez is a Fellow from Fundación Antorchas (Argentina).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W.A. Parrott
Rights and permissions
About this article
Cite this article
Rigano, M.M., Alvarez, M.L., Pinkhasov, J. et al. Production of a fusion protein consisting of the enterotoxigenic Escherichia coli heat-labile toxin B subunit and a tuberculosis antigen in Arabidopsis thaliana . Plant Cell Rep 22, 502–508 (2004). https://doi.org/10.1007/s00299-003-0718-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-003-0718-2